 Il mondo del
Tecnico Sanitario di Laboratorio Biomedico
Il mondo del
Tecnico Sanitario di Laboratorio Biomedico
Linee guida per la sicurezza e la salute dei lavoratori esposti ai chemioterapici antiblastici


Come vestirsi

La preparazione
NBP (Norme di Buona Preparazione)

Linee guida FU (Farmacopea Ufficiale)
Attenzione!!!! Come riportato dagli autori
Marcello Guidotti, Tommasina Coviello - copyright 2005-2006-2011-2013
questa pagina può essere riprodotta su qualsiasi supporto o rivista purché sia citata la fonte e
l'indirizzo di questo sito (ai sensi degli artt. 2575 e 2576 cc. Legislazione sul diritto d'autore). Le fotografie sono tratte da siti web e sono, o possono ritenersi, di pubblico dominio purché
utilizzate senza fini di lucro. Le immagini di prodotti presenti nel sito hanno unicamente valenza esemplificativa oltre che, eventualmente, illustrare messaggi fuorvianti e non vi è alcun
richiamo diretto o indiretto alla loro qualità e/o efficacia il cui controllo è affidato alle autorità regolamentatorie
Fonte: http://www.galenotech.org/galenici.htm
Trovi altro materiale interessante se vuoi approfondire ulteriormente
Grazie UNIROMA1 (proggetto didattico multimediale) Facoltà di medicina e farmacia
galenici magistrali, galenici officinali, farmaci generici
 La dizione "preparato galenico" deriva dal nome di Galeno, un medico dell'antica Grecia
che diffuse la pratica di comporre i rimedi medicamentosi miscelando varie sostanze di base. Le documentazioni della storia della farmacia testimoniano che sino a tutto il 19° secolo ed ai primi
trent'anni del 20°, la maggior parte dei rimedi venduti in farmacia venivano composti direttamente dal farmacista, conferendo così allo stesso la figura professionale di preparatore tecnico
piuttosto che dispensatore di farmaci.
La dizione "preparato galenico" deriva dal nome di Galeno, un medico dell'antica Grecia
che diffuse la pratica di comporre i rimedi medicamentosi miscelando varie sostanze di base. Le documentazioni della storia della farmacia testimoniano che sino a tutto il 19° secolo ed ai primi
trent'anni del 20°, la maggior parte dei rimedi venduti in farmacia venivano composti direttamente dal farmacista, conferendo così allo stesso la figura professionale di preparatore tecnico
piuttosto che dispensatore di farmaci.
Dopo la seconda guerra mondiale, con lo sviluppo dell'industrializzazione, la pratica della preparazione galenica, che da sempre costituiva l'aspetto caratterizzante la professione del
farmacista, ha subìto una progressiva riduzione sino a considerarsi residuale. In effetti, la possibilità di poter disporre con immediatezza di farmaci via via sempre più numerosi, tecnicamente
ineccepibili e terapeuticamente efficaci, realizzati da industrie farmaceutiche al passo con i progressi farmacologici e tecnico-farmaceutici ha indotto la classe medica a considerare
elettivamente la specialità medicinale.
Ciò nonostante, il farmacista ha conservato il suo ruolo di preparatore di prodotti medicinali, seppure nell'àmbito di particolari disposizioni legislative.
Il farmacista può ancóra disporre di un laboratorio, che è però obbligato per legge ad allestire con apparecchi, strumenti e sostanze indicati nella tabella 6 della Farmacopea Ufficiale. Il farmacista deve inoltre rifornirsi di tutti gli apparecchi, utensili, materiali, prodotti e reattivi adeguati al numero e alla natura delle preparazioni abitualmente eseguite nonché di idonee apparecchiature per il loro controllo come previsto dalle NBP (FU XI).
classificazione dei medicinali
Una volta ottenuta l'autorizzazione all'apertura di una farmacia, il farmacista è legittimato ad allestire
preparazioni estemporanee, classicamente note come "galenici magistrali" e "galenici officinali", i cosiddetti "multipli".
Prima di entrare nel dettaglio delle preparazioni estemporanee, per chiarezza è utile esaminare un breve
quadro riassuntivo per la classificazione dei medicinali:
-
medicinali di origine industriale

- specialità medicinali: preparati e messi in commercio con un nome fantasia (es. aspirina) e in confezioni particolari che non possono essere modificate dal farmacista. Necessitano di AP (Autorizzazione alla Produzione) e AIC (Autorizzazione Immissione in Commercio);
- medicinali equivalenti (ex generici): i medicinali non più protetti da brevetto possono essere preparati e commercializzati - dopo aver ottenuto l'AIC - con la denominazione comune della sostanza (es. acido acetilsalicilico) o, in sua assenza, con la denominazione scientifica (es. acido o-idrossibenzoico).
-
medicinali allestiti in farmacia

- galenici magistrali: medicinali destinati a un determinato paziente e preparati in farmacia su prescrizione medica;
- galenici ospedalieri: medicinali preparati nella farmacia ospedaliera e destinati a essere impiegati esclusivamente all'interno dell'ospedale;
- galenici multipli: medicinali preallestiti, preparati nella farmacia aperta al pubblico o in quella ospedaliera in base alle formulazioni presenti nel capitolo Preparazioni Farmaceutiche Specifiche della F.U. XI (precedentemente facenti parte del Formulario Nazionale). Queste preparazioni sono destinate ai clienti della farmacia o all'interno dell'ospedale. Il farmacista è sempre responsabile solo della qualità delle sostanze utilizzate e della corretta tecnica di preparazione.

Le formulazioni indicate nel Formulario Nazionale, non più compreso nella F.U. XI, possono comunque essere preparate in quanto sono riportate in un testo
ufficiale.
preparazioni con ricetta e senza ricetta
I galenici magistrali sono farmaci preparati dal farmacista in farmacia "secundum artem" su richiesta dei
fruitori che presentano una prescrizione del medico, il quale indicando espressamente qualità e quantità di ogni componente per adattare la formulazione alle specifiche necessità del suo
paziente, si assume le responsabilità relative all'efficacia e alla sicurezza della
formulazione.
La fonte di legittimazione dell'operato del farmacista è dunque rappresentata esclusivamente dalla ricetta
medica, poiché la formulazione quali-quantitativa non è codificata in alcun testo ufficialmente riconosciuto, ma è stabilita dal medico in funzione delle esigenze terapeutiche di ogni singolo
paziente (personalizzazione della terapia).
Il prodotto va allestito estemporaneamente, cioè al momento e pertanto non è lecita una preparazione
precedente alla prescrizione della ricetta medica.
Al farmacista compete il controllo della prescrizione medica per quanto riguarda esclusivamente gli aspetti tecnico-farmaceutici (compatibilità, dosaggio, ecc.) e legislativi (rispetto di specifiche norme, divieti, limitazioni, ecc.). Il farmacista è altresì responsabile della qualità delle sostanze utilizzate e della corretta tecnica di preparazione (le sostanze impiegate devono avere i requisiti prescritti nelle relative monografie riportate nella Farmacopea Ufficiale; le sostanze non iscritte in FU. devono essere comunque utilizzate dal farmacista allo stato di massima purezza, genuinità e ottima conservazione).
Le preparazioni galeniche possono essere allestite preventivamente nella quantità necessaria a soddisfare le esigenze della farmacia per essere destinate ai pazienti che si servono di tale esercizio (galenici multipli). La loro preparazione in farmacia, pertanto, è indipendente dalla ricetta medica, che disciplina invece la loro dispensazione al pubblico. In relazione al tipo di sostanze presenti nella formulazione, le preparazioni estemporanee possono essere infatti dispensate: senza ricetta medica; con ricetta medica ripetibile, non ripetibile, a ricalco.
Norme di Buona Preparazione
Le sostanze impiegate per le preparazioni galeniche devono avere caratteristiche corrispondenti a quelle riportate nella F.U. Il loro utilizzo è comunque subordinato alla verifica della prescrizione medica che il farmacista deve eseguire accertando l'esistenza dei requisiti sostanziali (compatibilità chimico-fisico-farmacologica, dosaggi) e formali previsti dalle normative vigenti.
Il farmacista che intenda allestire nella propria farmacia galenici officinali in forma multipla deve attenersi a quanto previsto nelle Norme di Buona Preparazione (N.B.P.) dei medicamenti in farmacia. Infatti l'allestimento deve avvenire attraverso procedure ben definite, che escludano possibilità di errore e che assicurano il possesso dei necessari requisiti di garanzia e omogeneità:
- è possibile preparare solo le formule indicate nel capitolo Preparazioni farmaceutiche specifiche della FU XI ed., secondo le previste modalità di allestimento;
- il laboratorio della farmacia deve rispondere ai requisiti previsti dalle Norme di Buona Preparazione (FU XI Ed. e decreti 19-11-03 e 22-06-05;
- sono allestibili solo quantitativi di prodotto adeguati alle necessità della propria farmacia e comunque non superiori ai 3 kg (FU XI Ed.);
- è necessario predisporre la documentazione tecnica relativa ad ogni preparazione allestita (lotto) per ciò che riguarda:
- le sostanze impiegate;
- le quantità prodotte;
- la procedura valida per la preparazione;
- il controllo di qualità eseguito sul prodotto finito;
- il prodotto deve essere etichettato adoperando la denominazione presente nel capitolo Preparazioni farmaceutiche specifiche della FU XI Ed. e adottando quanto previsto dalla N.B.P.;
- la preparazione deve essere etichettata e prezzata secondo le norme previste (Codice di autoregolamentazione);
- l'atto di dispensazione al pubblico deve essere accompagnato da dettagliate istruzioni sull'uso, indicando altresì le precauzioni da osservare, le eventuali indicazioni sull'eliminazione del contenitore e del residuo inutilizzato ed ogni altra informazione utile ad indirizzare il paziente ad un corretto utilizzo.
L'efficacia terapeutica di quanto prescritto dal medico esula dalle responsabilità del farmacista, il quale
però è unicamente e pienamente responsabile, per i galenici magistrali, sia delle proprietà sostanziali o intrinseche che di quelle formali o estrinseche. Questo impone al farmacista il controllo
di tutte le variabili che intervengono nell'allestimento del prodotto per rendere sempre riproducibile il processo di produzione, eliminando le possibilità di errore che potrebbero costituire
fattore di rischio per la salute del paziente.
Le variabili controllabili del medicamento sono il principio attivo, la formulazione e la tecnica di
allestimento. Ciascuno di questi elementi rappresenta un'area di rischio potenziale capace di compromettere l'efficacia, la sicurezza e la qualità del medicinale, cioè del prodotto o del
preparato destinato a correggere, modificare o ripristinare le condizioni organiche nell’uomo e nell'animale (secondo la definizione normativa di farmaco contenuta nel codice comunitario concernente i medicinali
per uso umano.
La Farmacopea indica i requisiti di qualità delle materie prime destinate ad essere impiegate nei medicinali, siano esse farmacologicamente attive o ausiliarie (additivi).
Le Norme di Buona Preparazione, sono invece contenute nella FU XI Ed. e codice comunitario concernente i medicinali per uso umano; esse sono un complesso di norme che tendono ad uniformare le
condizioni di preparazione in maniera tale da garantire in ogni luogo e in qualsiasi momento l'efficacia, la sicurezza, e la qualità del medicinale approntato. Queste norme coinvolgono il
personale, l'area delle preparazioni, le attrezzature di dotazione, le norme igieniche, le materie prime impiegate, il preparato galenico finito, l'etichettatura, il contenitore e i controlli da
effettuare sul preparato galenico finito, il lotto di preparazione del galenico e infine la procedura di autoispezione.
Analizzando le N.B.P., un problema di particolare delicatezza, che ha importanti riflessi sulla qualità dei prodotti allestiti, è costituito dalle misure igieniche e organizzative che devono essere assunte nel laboratorio galenico. Nell'àmbito dei locali della farmacia deve essere innanzitutto individuata un'area dove eseguire le preparazioni galeniche; lo spazio necessario dipende da che cosa e da quanto si prevede di produrre: quest'area deve essere isolata o isolabile dalle altre attività normalmente svolte in farrnacia. Un’area delle preparazioni deve essere dotata di un efficiente sistema di illuminazione, aspirazione e climatizzazione (temperatura e umidità). Le pareti e il soffitto devono essere lisci e rivestiti con materiale lavabile e inattaccabile da parte dei detergenti comunemente utilizzati.
L'organizzazione dell'area deve essere tale da garantire che non possa esistere commistione di materie prime con i prodotti finiti, che devono essere lavorati e conservati separatamente; è altresì indispensabile evitare la contaminazione crociata delle sostanze farmaceutiche con altre sostanze farmaceutiche di tipo diverso.
(modificato e aggiornato da Acta Phytotherapeutica)
vincoli del medico per la prescrizione
Tra le ragioni per cui un medico ricorre a preparazioni estemporanee, c'è l'esigenza di utilizzare particolari preparazioni non disponibili nella corrente produzione industriale che, per il tipo di formulazione qualitativa e quantitativa sono ritenute necessarie e/o talvolta insostituibili per soddisfare protocolli di terapia messi a punto per patologie che prevedono trattamenti personalizzati, elaborati in funzione di numerosi parametri che generalmente sono molto variabili da paziente a paziente.
La terapia del dolore è tra quelle che si avvale di trattamenti farmacologici "personalizzati". Questi trattamenti sono efficacemente ottenuti mediante preparazioni estemporanee, frutto di un
rapporto di costante collaborazione professionale tra medico e farmacista.
Considerato che il dolore è definito "una sgradevole esperienza sensoriale ed emotiva associata ad un effettivo o potenziale danno tissutale... e che dunque il dolore è sempre una esperienza
soggettiva" si può facilmente comprendere come il ricorso ad una terapia farmacologica personalizzata sia di fondamentale importanza.
I medici possono prescrivere preparazioni magistrali esclusivamente a base di princìpi attivi descritti nelle Farmacopee in vigore nei Paesi dell'Unione Europea o contenuti in medicinali prodotti industrialmente di cui è autorizzato il commercio in Italia o in altro Paese dell'Unione europea.
Con il DL 17.02.1998 (convertito in Legge n. 94 dell’8.4.1998), la prescrizione di preparazioni magistrali per uso orale è stata ampliata includendo princìpi attivi diversi da quelli previsti dal punto precedente, qualora questi siano contenuti in prodotti nonfarmaceutici per uso orale, regolarmente in commercio nei Paesi dell'Unione europea.
La prescrizione di preparazioni magistrali per uso esterno può includere princìpi attivi diversi da quelli previsti al primo punto, qualora questi siano contenuti in prodotti cosmetici regolarmente in commercio nei Paesi dell'Unione europea (es., acido glicolico).
E' consentita la prescrizione di preparazioni magistrali a base di princìpi attivi già contenuti in specialità medicinali la cui autorizzazione all'immissione in commercio sia stata revocata o non confermata per motivi non attinenti ai rischi di impiego del principio attivo (es., revoca dovuta a insufficiente standard qualitativo della produzione).
Prima dell’esecuzione della preparazione il farmacista deve accertarsi che le dosi prescritte non superino quelle massime consentite dalla Tabella 8 della FU. La prescrizione di sostanze in dosi eccedenti quelle previste è consentita a condizione che il medico sottoscriva espressamente l'assunzione di responsabilità indicando altresì l'uso farmacologico cui la preparazione è destinata (artt. 34, 40 R.D. 1706/38).
Se la prescrizione è effettuata per indicazioni
corrispondenti a quelle delle specialità in commercio contenenti lo stesso principio attivo, la ricetta non deve contenere particolari dichiarazioni da parte del medico. Al contrario, se è
rilasciata per indicazioni terapeutiche diverse, il medico deve dichiarare di aver ottenuto il consenso del paziente, specificare le esigenze particolari che giustifichino il ricorso alla
prescrizione estemporanea e utilizzare un codice di riferimento numerico o alfanumerico di collegamento a dati di archivio in suo possesso che consenta, in caso di richiesta da parte
dell'Autorità sanitaria, di risalire all'identità del paziente.
Le ricette che riportano i dati di cui sopra (consenso, codice ecc.) devono essere trasmesse mensilmente dal farmacista, in originale o in copia, all'Azienda ASL per il successivo inoltro al Ministero della Salute.
vincoli del farmacista per la preparazione
Secondo una nota della FOFI inviata al Ministero della Sanità (febbraio 1998):
- le preparazioni galeniche magistrali possono essere eseguite esclusivamente su prescrizione medica;
- sussiste il divieto assoluto di procedere alla preparazione prima della presentazione della ricetta medica.
- sussiste l'obbligo di uniformarsi integralmente alla prescrizione medica, e quindi di rispettare assolutamente la quantità, il dosaggio e la forma farmaceutica prescritti dal medico;
- sussiste l'assoluto divieto di sostituire una specialità medicinale, prescritta dal medico nella ricetta, con una preparazione galenica, anche se quest'ultima fosse a base del medesimo principio attivo e avesse dosaggio, forma farmaceutica e via di somministrazione identici alla specialità;
- sussiste l'assoluto divieto di utilizzare prodotti già preparati o anche semilavorati o di sconfezionare specialità medicinali, per eseguire la preparazione;
- la preparazione deve essere eseguita integralmente nella farmacia nella quale viene poi venduta; non è pertanto consentito affidare l'esecuzione della preparazione, in tutto in parte, ad altre farmacie o a laboratori esterni.
Sulla base di questa nota, si evince che la legittimazione di una preparazione magistrale risiede o nella ricetta medica (o medico-veterinaria) o nel capitolo Preparazioni Farmaceutiche Specifiche della FU XI Ed.; in quest'ultimo caso la Farmacia può allestire preparazioni sotto forma di multipli.
Nell'allestimento di preparazioni magistrali assumono particolare importanza le disposizioni contenute nelle Tabelle 4 e 5 della FU ai fini della ripetibilità o meno della ricetta.
La strumentazione di cui una farmacia deve essere obbligatoriamente provvista è riportata nella Tabella 6 della FU.
Le ricette che riportano dati sensibili (consenso, codice ecc.) devono essere trasmesse mensilmente dal farmacista, in originale o in copia, all'Azienda ASL per il successivo inoltro al Ministero della Salute.
preparazioni estemporanee contenenti princìpi attivi coperti da brevetto
Negli ultimi anni è divenuta pratica corrente la prescizione di preparazioni galeniche a base di princìpi attivi coperti da brevetto. Questa pratica trova giustificazione nel fatto che le preparazioni così allestite hanno un prezzo calcolato secondo la Tariffa Nazionale dei Medicinai, inferiore rispetto a quello delle specialità medicinali (v. per es. le capsule a base di chitosano). Tale pratica, il cui contenzioso si basa sulla cosiddetta "eccezione galenica" ha spinto le aziende farmaceutiche detentrici del brevetto ad avviare azioni legali che hanno detrminato una nuova disciplina (v.).
preparazioni magistrali contenenti sostanze dopanti
Le preparazioni galeniche, officinali o magistrali che contengono principi attivi o eccipienti appartenenti alle classi farmacologicamente vietate in quanto considerate doping sono prescrivibili solo dietro presentazione di ricetta medica non ripetibile. Il farmacista è tenuto a conservare l'originale della ricetta per 6 mesi. (art. 7 ,c.4 -L.14.12.2000, N. 376 "Disciplina della tutela sanitaria delle attività sportive e della lotta contro il doping", GU 18.12.2000, n. 294).
Il DECRETO 24 settembre 2003 del Ministero della Salute (G.U. n. 257 del 5-11-2003) ha fissato le Modalità di attuazione delle disposizioni contenute nell'art. 7 della legge 14 dicembre 2000, n. 376, recante «Disciplina della tutela sanitaria delle attività sportive e della lotta contro il doping».
tariffazione ed etichettatura delle preparazioni estemporanee
Il farmacista ha l'obbligo di realizzare la preparazione galenica magistrale nel più breve tempo possibile, compatibilmente con i tempi richiesti per il reperimento delle sostanze nel caso ne sia sprovvisto.
Il prezzo delle preparazioni magistrali è calcolato secondo le norme contenute nella Tariffa Nazionale dei Medicinali, TN. Nel caso che una sostanza non sia presente nella TN il prezzo si determina raddoppiando quello di acquisto al netto dell'IVA e addizionandovi l'aliquota IVA relativa al prodotto finito (10% nel caso dei medicinali).
Es. se la farmacia ha acquistato una sostanza non inclusa nella TN al prezzo di 0,5 € al grammo (+ IVA 10%), in una preparazione che ne contenga 1 g il prezzo di vendita relativo a questa sostanza sarà 0,5 € · 2 = 1 € + 0.5 · 10% (aliquota IVA sul medicinale) = 1,05 €.
Per quanto riguarda l'etichettatura delle preparazioni magistrali (art. 37 RD 30.09.1938, n. 1706), il farmacista ha l'obbligo di indicare sui recipienti e sugli involucri dei medicinali da lui allestiti in farmacia:
- la data di spedizione;
- l’indicazione quali-quantitativa del rimedio secondo la ricetta e senza utilizzare formule chimiche;
- la dose da somministrare;
- la via di somministrazione;
- il prezzo praticato, indicando specificamente:
- l’importo complessivo delle sostanze;
- l’importo complessivo degli onorari professionali;
- il costo del recipiente, quando sia fornito dal farmacista.
- l'eventuale presenza di sostanze velenose deve essere evidenziata in modo visibile.
E' inoltre opportuno indicare il nome del paziente e del medico prescrittore.
Per quanto attiene l'etichettatura di multipli (N.B.P. FU IX ed), l’etichetta deve riportare:
- nome, indirizzo, numero di telefono della Farmacia;
- data di preparazione;
- numero identificativo del lotto;
- composizione quali-quantitativa della preparazione, quale risulta nella formulazione originale, integrata dalle sostanze eventualmente aggiunte per motivi tecnici e presenti nel preparato (es. non è sufficiente indicare: Eccipiente q. b. ma si dovranno indicare natura e quantità dei singoli eccipienti);
- le indicazioni addizionali, se il medicinale è soggetto a particolare disciplina;
- la data entro la quale il medicinale deve essere utilizzato;
- ogni altra indicazione prevista dalle leggi e dai regolamenti.
Tutte le ricette galeniche magistrali, ripetibili o non
ripetibili, hanno validità 3 mesi, ad eccezione delle prescrizioni di stupefacenti di II tabella, la cui validità è sempre di 30 giorni. Anche se è invalsa nell'uso, l'apposizione del timbro
della farmacia non è prevista da alcuna normativa relativa a galenici magistrali, sebbene riaffermata dal Ministero della Salute con proprie note. Il farmacista ha inoltre l’obbligo di conservare
per 6 mesi le ricette spedite concernenti preparazioni estemporanee (art. 87 comma 7 Legge Finanziaria 2001). Tale periodo è esteso a 5 anni nel caso di preparazioni magistrali contenenti
sostanze stupefacenti di cui occorre documentare la movimentazione nel registro entrata-uscita.
Se il medico indica nella preparazione l’impiego di alcool denaturato, il farmacista è tenuto a sostituirlo con alcool etilico 96, in quanto è fatto divieto di impiegare alcoli diversi nelle
preparazioni farmaceutiche.
| Controlli da effettuare sulla preparazione | creme | monodose | soluzioni | emulsioni |
| Verifica della correttezza delle procedure eseguite | sì | sì | sì | sì |
| Controllo dell’aspetto | sì | sì | sì | sì |
| Controllo del confezionamento e in particolare della sua tenuta | sì | sì | sì | sì |
| Verifica della corretta compilazione dell’etichetta compresa l’indicazione delle modalità di conservazione e di vendita. | sì | sì | sì | sì |
| L’uniformità di massa che deve essere accertata su un campione la cui dimensione dipende dalla consistenza numerica delle dosi forma. Nessuna dose forma del campione dovrà discostarsi dal ± 10 per cento del peso medio.Nel caso delle capsule, il controllo dell’uniformità di massa si effettuerà sulle capsule piene. | no | sì | no | no |
| la quantità o il numero di dosi forma da dispensare. | no | sì | no | no |
| l’aspetto e l’assenza di particelle visibili a occhio nudo, | no | no | sì | no |
| il pH, se necessario. | no | no | sì | no |
| l’aspetto della preparazione | no | no | no | sì |
| la ridispersibilità delle fasi. | no | no | no | sì |
conservazione delle sostanze
Le materie prime devono essere conservate in contenitori inerti e muniti di adatta chiusura, le cui caratteristiche sono riportate in Farmacopea. Tutti i contenitori devono essere coerentemente etichettati con l'indicazione della sostanza.
Per "Recipiente ben chiuso" si intende un contenitore in grado di proteggere il contenuto da inquinamento per solidi o liquidi provenienti dall’esterno, conservando il medicamento praticamente inalterato nelle ordinarie condizioni d’uso, di conservazione, di trasporto e di ambiente. In questo senso sacchetti in carta opaca, politenati, chiusi con risvolti della parte superiore sono idonei a contenere la maggior parte delle polveri non igroscopiche.
Non è corretto trasferire materie prime dal contenitore fornito dal produttore (che deve essere del tipo conforme al requisito FU) in altri contenitori, in quanto possono verificarsi problemi di inquinamento, perdita dei dati del lotto, perdita dei dati analitici o delle specifiche tecniche del prodotto ricevuto.
Il concetto di scadenza - inteso come data limite oltre la quale un medicinale non può essere utilizzato - . Sebbene esistano sostanze inorganiche la cui stabilità, se conservate in condizioni idonee, è "illimitata" (es. iodio, sodio cloruro, calcio carbonato, argento nitrato ecc.), è invalsa la procedura di eliminare sostanze "prodotte" o riconfezionate da un distributore da oltre 5 anni; tuttavia questa accortezza non è di per sé sufficiente a garantire la purezza del prodotto impiegato.
E’ inoltre necessario seguire semplici criteri:
- eliminare tutte le sostanze visibilmente alterate (cambi di colore, di odore, di aspetto)
- eliminare le sostanze che non vengono più utilizzate per preparazioni galeniche magistrali, salvo che non si tratti di sostanze obbligatorie.
- eseguire un controllo del punto di fusione in caso di sostanze organiche con punto di fusione definito.
Non è possibile stabilire con certezza un periodo di validità per le preparazioni galeniche estemporanee, destinate per loro natura ad essere utilizzate entro breve tempo dall’acquisto;
tuttavia in etichetta, deve essere indicata la data di scadenza che può superare un massimo di sei mesi dalla data di preparazione.
In particolare, per quanto attiene le NPB, secondo la F.U. XI, in assenza di informazioni sulla stabilità devono essere osservati, per preparati non sterili, i seguenti limiti di utilizzazione
della preparazione stessa conservata nelle condizioni indicate in etichetta.
-
formulazioni solide, liquide non acquose o con un contenuto alcoolico non inferiore al 25 per cento: non oltre il 25 per cento del più breve periodo di validità dei
componenti utilizzati; tale periodo non può comunque superare i 6 mesi.
- per tutte le altre formulazioni: utilizzare entro 30 giorni dalla data di preparazione. Questo limite deve essere ridotto o può essere superato solo sulla base di specifiche conoscenze ed accorgimenti connessi con la contaminazione microbica del preparato e con le caratteristiche chimico-fisiche dei suoi componenti.
preparazioni galeniche allestibili nelle parafarmacie
Con l'articolo 11 (comma 17) del Decreto-Legge del 24 gennaio 2012 convertito con modificazioni dalla L. 24 marzo 2012, n. 27 (in S.O. n. 53, relativo alla G.U. 24/03/2012, n. 71) , è stata data alle parafarmacie, corner, ecc., la possibilità di allestire preparazioni galeniche officinali; in particolare :
17. "Gli esercizi commerciali di cui all'articolo 5, comma 1, del decreto-legge 4 luglio 2006, n. 223, convertito, con modificazioni, dalla legge 4 agosto 2006, n. 248, in possesso dei requisiti vigenti [si tratta di parafarmacie, corner, ecc. - NdR], sono autorizzati, sulla base dei requisiti prescritti dal decreto ministeriale previsto dall'articolo 32, comma 1, del decreto legge 6 dicembre 2011, n. 201, convertito con modificazioni dalla legge 22 dicembre 2011, n. 214, ad allestire preparazioni galeniche officinali che non prevedono la presentazione di ricetta medica, anche in multipli, in base a quanto previsto nella farmacopea ufficiale italiana o nella farmacopea europea."
attività critiche: attività che hanno influenza sulla qualità della preparazione fornita al paziente
farmacopea in vigore: Farmacopea Ufficiale della Repubblica Italiana (costituita dalla XI edizione della F.U. e dalla V edizione della Farmacopea Europea, recepita
direttamente in lingua inglese e francese, con i suoi futuri supplementi quadrimestrali) e tutte le farmacopee nazionali in vigore negli Stati Membri dell’Unione Europea.
limiti di accettabilità: intervallo numerico entro il quale dovrà situarsi il risultato di un eventuale dosaggio quantitativo del/i principio/i attivo/i presente/i nel
preparato. I limiti di accettabilità sono fissati in base a: composizione del preparato; procedura di preparazione; metodo utilizzato per un eventuale controllo del preparato; processo di
degradazione del principio attivo nel tempo di utilizzazione del preparato.
preparato magistrale o formula magistrale: preparazione allestita in farmacia in base ad una prescrizione medica destinata ad un determinato paziente. Sono tecnicamente
assimilabili ai preparati magistrali anche tutte le miscelazioni, diluizioni, ripartizioni, ecc., eseguite per il singolo paziente su indicazione medica. La prescrizione medica deve tenere conto
di quanto previsto dall'articolo 5 del decreto legge 17 febbraio 1998, n. 23 , convertito in legge con modificazioni dall'articolo 1, comma 1, legge 8 aprile 1998, n.94. (v. riquadro
avanti)
preparato officinale o formula officinale: preparazione allestita in farmacia in base alle indicazioni di una farmacopea e destinata ad essere fornita direttamente ai
pazienti che si servono in tale farmacia.
responsabile: ha il compito di eseguire e/o controllare un'attività. Secondo la tipologia e il carico di lavoro della farmacia, possono esistere vari livelli di
responsabilità, in cui i gradi inferiori fanno comunque capo al responsabile generale (farmacista titolare o direttore).
scala ridotta: numero di preparazioni pronte per la vendita eseguibili dal farmacista. La consistenza numerica, compatibilmente con la stabilità del preparato stesso, è
quella ottenibile da una massa non più grande di 3000 grammi di formulato. Per i preparati soggetti a presentazione di ricetta medica la consistenza numerica deve essere documentata sulla base
delle ricette mediche (copie e originali) presentate dai pazienti. Il farmacista può procedere ad una successiva preparazione di una certa formula officinale purché non superi la consistenza
numerica prevista dalla scala ridotta.
|
Legge 8 aprile n.94
Art. 5.
Prescrizione di preparazioni magistrali 1. Fatto salvo il disposto del comma 2, i medici possono prescrivere preparazioni magistrali esclusivamente a base di principi attivi descritti nelle farmacopee dei Paesi dell'Unione europea o contenuti in medicinali prodotti industrialmente di cui è autorizzato il commercio in Italia o in altro Paese dell'Unione europea. La prescrizione di preparazioni magistrali per uso orale puo' includere principi attivi diversi da quelli previsti dal primo periodo del presente comma, qualora questi siano contenuti in prodotti non farmaceutici per uso orale, regolarmente in commercio nei Paesi dell'Unione europea; parimenti, la prescrizione di preparazioni magistrali per uso esterno puo' includere principi attivi diversi da quelli previsti dal primo periodo del presente comma, qualora questi siano contenuti in prodotti cosmetici regolarmente in commercio in detti Paesi. Sono fatti in ogni caso salvi i divieti e le limitazioni stabiliti dal Ministero della sanita' per esigenze di tutela della salute pubblica. 2. E' consentita la prescrizione di preparazioni magistrali a base di principi attivi già contenuti in specialità medicinali la cui autorizzazione all'immissione in commercio sia stata revocata o non confermata per motivi non attinenti ai rischi di impiego del principio attivo. 3. Il medico deve ottenere il consenso del paziente al trattamento medico e specificare nella ricetta le esigenze particolari che giustificano il ricorso alla prescrizione estemporanea. Nella ricetta il medico dovrà trascrivere, senza riportare le generalità del paziente, un riferimento numerico o alfanumerico di collegamento a dati d'archivio in proprio possesso che consenta, in caso di richiesta da parte dell'autorità sanitaria, di risalire all'identità del paziente trattato. 4. Le ricette di cui al comma 3, in originale o in copia, sono trasmesse mensilmente dal farmacista all'azienda unità sanitaria locale o all'azienda ospedaliera, che le inoltrano al Ministero della sanità per le opportune verifiche, [anche ai fini dell'eventuale applicazione dell'articolo 25, comma 8, del Dlgs 29 maggio 1991, n. 178 - Decreto abrogato e sostituito da "codice comunitario concernente i medicinali per uso umano" cfr. TITOLO IX, Farmacovigilanza]. 5. Le disposizioni dei commi 3 e 4 non si applicano quando il medicinale è prescritto per indicazioni terapeutiche corrispondenti a quelle dei medicinali industriali autorizzati a base dello stesso principio attivo. 6. La violazione, da parte del medico o del farmacista, delle disposizioni del presente articolo è oggetto di procedimento disciplinare ai sensi del decreto legislativo del Capo provvisorio dello Stato 13 settembre 1946, n. 233. |
galenici rimborsabili: il modello Bolzano
Nella provincia di Bolzano le preparazioni galeniche magistrali, prescritte da medici convenzionati e allestite in farmacia, vengono rimborsate dal Servizio sanitario provinciale in virtù della legge provinciale n 2 del 3 gennaio1986 e successive modifiche, e in base al regolamento approvato con propria deliberazione n. 643 del 10 marzo 2003. «Per comprendere questa norma» ha spiegato Luca Collerata, presidente di Federfarma Bolzano, a Fitoterapia33 «occorre una premessa storica». La forte tradizione di preparatori tipica dell'area austriaca, infatti, ha portato, circa 8 anni fa, Federfarma Bolzano, in collaborazione con il professor Cini dell'università di Bologna, a elaborare un Formulario provinciale galenico in parallelo a quello Nazionale. Quindi la norma provinciale di fatto ribadisce il diritto del paziente a vedersi rimborsato anche nel caso di medicinali magistrali allestiti in farmacia secondo le regole della Farmacopea ufficiale. Il regolamento provinciale originario ha subìto poi nel tempo diverse modifiche l'ultima delle quali, approvata con delibera n. 956 del 30.03.2009, prevede l'erogazione a carico dell'Ssp delle sole preparazioni galeniche magistrali a base di sostanze iscritte nell'allegato A, con alcune eccezioni. «Ad esempio» specifica Collerata a Fitoterapia 33 «i rimborsi non sono previsti per i farmaci per i quali esista in commercio una specialità con composizione uguale per dosaggio e forma farmaceutica e con prezzo inferiore. Sono escluse anche la preparazioni iniettabili, gli omeopatici, le tisane, gli elixir e i vini medicati». È invece previsto, su prescrizione di uno specialistica, il rimborso di magistrali che prevedono lo sconfezionamento di medicinali industriali*, comprese le specialità medicinali. «Per ogni singola preparazione galenica» aggiunge il presidente di Federfarma Bolzano «l'assistito è tenuto, a titolo di partecipazione alla spesa farmaceutica, al pagamento del 50% del prezzo di vendita al pubblico, con arrotondamento ai ¬0,25 superiori». Questa norma presenta quindi una duplice valenza positiva: oltre a consentire ai pazienti di disporre di farmaci personalizzati rimborsati dalla parte pubblica, riconosce e legittima una delle attività più qualificanti della professione farmaceutica. (Farmacista 33 - 19 settembre 2011)
* L'obbligo di attenersi a quanto prescritto dal medico trova legittimo ostacolo nella sola ipotesi in cui il farmacista individui, nella ricetta, la prescrizione di sostanze velenose a dosi non medicamentose o pericolose, dovendo in tal caso esigere (ex art. 40 del regolamento per il servizio farmaceutico n. 1706 del 1938) che il medico «dichiari per iscritto che la somministrazione avviene sotto la sua responsabilità, previa indicazione dello scopo terapeutico perseguito» [Cassazione civile, sez. III, sentenza 28.03.2008 n. 807]. Ne discende che è ragionevole supporre che, in caso di sconfezionamento del medicinale, la responsabilità di eventuali effetti avversi o interefernze tra eccipienti sia riconducibile al medico prescrittore (NdR).
polveri
La più semplice forma farmaceutica è costituita dalle polveri, galeniche o industriali che devono soddisfare specifici requisiti indicati dalla F.U.
La formulazione di farmaci in polveri, per impieghi non aspersori, è a volte preferibile rispetto alle compresse sia per la possibilità di adattare meglio la dose al paziente, sia per la maggior
biodisponibilità dovuta all'eliminazione della fase di disaggregazione della compressa. La superficie specifica delle particelle che costituiscono la polvere, influenza la velocità di dissoluzione o addirittura l'attività nel caso di sostanze poco solubili.
Durante il mescolamento di due polveri, si possono formare
miscele eutettiche, caratterizzate da un punto di fusione inferiore a quello dei singoli componenti con possibile rammollimento e liquefazione della miscela a temperatura ambiente.
Questo fenomeno, che può verificarsi anche dopo un certo tempo, si può spiegare considerando che la miscela di due polveri fa sì che venga reciprocamente ridotta la simmetria dei loro reticoli
cristallini, con riduzione delle forze intermolecolari e conseguente abbassamento del punto di fusione.
La lavorazione di una preparazione eutettica comporta difficoltà sia a livello galenico che industriale. Un metodo per evitare il problema consiste nel mescolare le polveri, o preferibilmente i
cristalli, senza triturarle, in modo da ridurne la superficie di contatto. Si può anche usare un un terzo componente (ad es. caolino, talco, ecc.) che, miscelato al primo, agendo da barriera
protettiva limita il contatto tra le polveri eutettiche; oppure lasciare rammollire le polveri e quindi correggerne la consistenza facendole adsorbire su talco o caolino.
Qualora questi accorgimenti, particolarmente a livello galenico, si dimostrassero inadeguati, occorrerà separare i componenti in due cartine o cialde, indicando nell'etichetta della confezione
che le cartine o cialde "A" e "B" devono essere riunite al momento dell'uso.
Da quanto discusso, segue che il punto di fusione di una
polvere può essere assunto quale criterio di purezza, in quanto eventuali particelle estranee ne abbassano il valore. A rigore, l'abbassamento del punto di fusione si verifica solo in un sistema
ideale in cui, per definizione, i due solidi siano solubili allo stato fuso (es., le coppie metalliche Al-Sn, Si-Al, Ob-Ag, i sali fusi tipo KCl-AgCl, o sostanze organiche come
cicloesano-benzene).
analisi termica di miscele
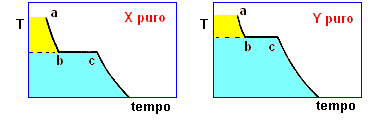 la curva di raffreddamento di X puro (prima figura a
sinistra) presenta una diminuzione di temperatura regolare dal punto (a) al punto (b) in accordo con il fatto che diminuisce l'energia cinetica della sostanza. A partire dal punto (b), ha inizio
la fase di cristallizzazione, durante la quale la temperatura resta costante. Questo perché le particelle, sempre più lente per la diminuzione della temperatura, si assestano nella loro matrice
cristallina e durante la formazione dei legami liberano energia in misura tale da compensare il calore che viene sottratto dal sistema di raffreddamento esterno: la lunghezza del tratto (b-c) è
legata al calore latente (proporzionale all'intensità delle forze di legame presenti nel solido puro) tipico della transizione di fase.
la curva di raffreddamento di X puro (prima figura a
sinistra) presenta una diminuzione di temperatura regolare dal punto (a) al punto (b) in accordo con il fatto che diminuisce l'energia cinetica della sostanza. A partire dal punto (b), ha inizio
la fase di cristallizzazione, durante la quale la temperatura resta costante. Questo perché le particelle, sempre più lente per la diminuzione della temperatura, si assestano nella loro matrice
cristallina e durante la formazione dei legami liberano energia in misura tale da compensare il calore che viene sottratto dal sistema di raffreddamento esterno: la lunghezza del tratto (b-c) è
legata al calore latente (proporzionale all'intensità delle forze di legame presenti nel solido puro) tipico della transizione di fase.
Completata la fase di cristallizzazione, la temperatura di X prende nuovamente a diminuire a séguito della cessione di calore al sistema di raffreddamento.
La curva di raffreddamento di Y puro può essere discussa come per X puro
I diagrammi seguenti, si riferiscono all'analisi termica di due sostanze X ed Y con Tx < Ty.
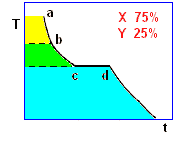 La curva di raffreddamento di una ipotetica
miscela composta da X(75%) e Y(25%), presenta tre punti singolari: (b), (c), (d). In particolare, da (a) a (b) si avrà un raffreddamento regolare della miscela finché, in corrispondenza di (b),
la cristallizzazione di Y (con maggior temperatura di fusione) comporta arresto termico per tutta la percentuale di questo componente, mentre X continua a raffreddare, però con pendenza diversa,
fino al punto (c), corrispondente alla temperatura di equilibrio, Te, dove tutta la massa di X inizia a cristallizzare.
La curva di raffreddamento di una ipotetica
miscela composta da X(75%) e Y(25%), presenta tre punti singolari: (b), (c), (d). In particolare, da (a) a (b) si avrà un raffreddamento regolare della miscela finché, in corrispondenza di (b),
la cristallizzazione di Y (con maggior temperatura di fusione) comporta arresto termico per tutta la percentuale di questo componente, mentre X continua a raffreddare, però con pendenza diversa,
fino al punto (c), corrispondente alla temperatura di equilibrio, Te, dove tutta la massa di X inizia a cristallizzare.
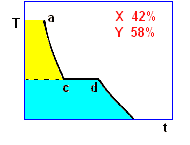 la curva di raffreddamento di una miscela composta
da X(42%) e Y(58%), presenta solo due punti singolari: (c), (d). Infatti, diminuendo la percentuale di X, il punto singolare (b) corrispondente al punto di fusione di X (nel grafico precedente),
si sposta fino a coincidere (per la particolare composizione ipotizzata) con il punto (c): in questo caso, abbiamo quella che prende il nome di miscela eutettica, caratterizzata da un unico punto
di fusione.
la curva di raffreddamento di una miscela composta
da X(42%) e Y(58%), presenta solo due punti singolari: (c), (d). Infatti, diminuendo la percentuale di X, il punto singolare (b) corrispondente al punto di fusione di X (nel grafico precedente),
si sposta fino a coincidere (per la particolare composizione ipotizzata) con il punto (c): in questo caso, abbiamo quella che prende il nome di miscela eutettica, caratterizzata da un unico punto
di fusione.
In genere le strutture con bassi valori di DH e t.f. (canfora, aspirina, ecc.), presentano più marcatamente i fenomeni esaminati. D'altra parte,
alcune sostanze mescolate insieme non formano una miscela bassofondente, il cui innesco, però, può essere indotto da un terzo componente: un esempio tipico è offerto dalla miscela fenile
salicilato-acetanilide-antipirina.
Fra le sostanze utilizzate per la preparazione di polveri galeniche, quelle che possono formare eutettici sono: acetanilide, acido acetilsalicilico, cloralio idrato, fenacetina, mentolo, fenolo,
fenile salicilato (salolo), timolo. Anche le miscele con p.f. dell'eutettico relativamente alto (81-90 oC), possono fondere a temperatura ambiente in quanto, triturandole nel
mortaio, si creano attriti che possono produrre, anche solo in alcune zone della miscela, notevoli aumenti della temperatura.
La tabella seguente, riporta alcune miscele eutettiche.
| salolo (41-43 oC) | + | canfora (164-179oC) | > 10oC |
| salolo (41-43oC) | + | timolo(51oC) | > 13oC |
| resorcina (110 oC) | + | acetanilide(164-179oC) | > 33oC |
| pirocatechina (104 oC) | + | acetanilide(164-179oC) | > 37oC |
| ac. acetilsalicilico (103-106oC) | + | acetanilide(164-179oC) | > 81oC |
| fenacetina (137 oC) | + | acetanilide(164-179oC) | > 90oC |
Altre caratteristiche delle polveri sono:
- igroscopicità: tendenza ad assorbire l'umidità dell'aria;
- deliquescenza: il solido si solubilizza nella propria acqua di cristallizzazione senza ssorbirla dall'esterno;
- efflorescenza: perdita di acqua di cristallizzazione (si verifica, per es. con ac. citrico, allume, atropina solfato, caffeina, calcio lattato, codeina fosfato, chinina, terpina idrata, ecc).
Per polverizzare le sostanze che si lavorano difficilmente, si ricorre alla cosiddetta "polverizzazione per intermedio". Per es., la canfora si deforma senza polverizzarsi, per cui la si solubilizza triturandola in un solvente volatile, in modo da ottenere un precipitato polveroso dopo la sua evaporazione. Un altro esempio: il salolo, sottoposto a triturazione, si elettrizza; così, per evitare che le sue particelle -omogeneamente cariche- si respingano spargendosi tutt'intorno, si aggiunge un agente coibente, tipo ligroina, che ne attenua gli effetti elettrostatici.
| mentolo racemico | 1 g |
| talco mentolato q.b a | 100 g |
Una classica preparazione è il mentolo polvere cutanea noto come talco mentolato. Secondo la F.U. XI è una polvere all'1% in mentolo racemico. Si prepara disperdendo il mentolo nel talco aggiunto per diluizioni progressive.
| mentolo | 1 g |
| canfora | 1 g |
| talco | 98 g |
Un'altra comune preparazione è il talco mento-canforato riportato in F.U. VI che essendo costituito da una miscela eutettica, richiede alcune delle accortezze citate per queste preparazioni.
Tipicamente si addiziona talco e mentolo e si aggiunge con diluizioni progressive il talco in modo da estinguere la massa deliquescente.
polvere di Dover
 Cephaelis ipecacuanha |
Thomas Dover ("medico" e bucaniere inglese, 1660-1742), nel 1710 elaborò un preparato contro la gotta a base d'oppio, liquirizia, salnitro e ipecacuana: la polvere di Dover, che divenne uno dei
farmaci più usati del XVII secolo. La polvere di Dover andava sciolta in un bicchiere di latte caldo cagliato con vino bianco e presa prima di andare a letto. «Coprendosi bene e bevendone dalle
due alle tre pinte, in modo da sudare molto, in due o tre ore al massimo, il paziente non avvertirà più il dolore», assicurava la formula illustrativa scritta da Dover stesso.
Quando Dover utilizzò dosaggi maggiori, provocò molti problemi, allontanando i medici dall'uso dell'oppio. Così, agli inizi dell'800, i medici adottarono il criterio che «se il paziente si
lamenta per i dolori, lasciatelo lamentare. I lamenti non hanno mai ucciso nessuno...»1. Il trattamento del dolore era diventato pericoloso.
La formulazione corrente della polvere di Dover prevede 10 parti di polvere di oppio, 10 parti di polvere di ipecacuana e 80 parti di lattosio (questa preparazione non può essere allestita in
quanto l'oppio è unicamente contenuto nella tab. I degli stupefacenti).
La polvere di oppio, in questo caso, ha azione di tipo morfinico ma la presenza dell'ipecacuana non permette di abusarne in quanto quest'ultima droga contiene emetina che a forti concentrazioni
induce il vomito. La polvere di Dover contiene l'1% di morfina in quanto la polvere di oppio F.U. contiene il 10% di morfina.
1Abbey Strauss - The Pain Project Volume 2, Issue 1 January 1994
sacchetti refrigeranti
Le miscele eutettiche racchiuse in piccoli involucri sono utilizzate per mantenere valori di bassa temperatura all'interno di un contenitore termico senza utilizzare macchine frigorifere. Queste
miscele si ottengono sfruttando soluzioni acquose di opportuni sali.
Un altro impiego comune si ha per i refrigeranti gengivari usati per alleviare i dolori gengivari durante la dentizione dei bambini.
Il principio è il seguente: a temperatura ambiente ed a pressione atmosferica costante, si miscela del ghiaccio tritato con un sale, ad esempio, cloruro di sodio in proporzioni tali da ottenere
un sistema formato da due fasi solide (il ghiaccio e il sale) ed una liquida (la soluzione acquosa di cloruro di sodio).
Poiché queste tre fasi possono coesistere solamente al punto eutettico (in questo caso vale - 21,3 °C), a temperatura ambiente non si ha equilibrio e quindi il ghiaccio fonde e viene
solubilizzato altro cloruro di sodio. Poiché il processo di fusione richiede assorbimento di calore dall'ambiente esterno, si ha un conseguente effetto refrigerante.
 Mescolando opportune quantità di ghiaccio e sale è possibile
formare miscele frigorifere efficienti (ricaricabili mettendole in frigorifero per alcune ore) e con una buona durata nel tempo (arco di ore).
Mescolando opportune quantità di ghiaccio e sale è possibile
formare miscele frigorifere efficienti (ricaricabili mettendole in frigorifero per alcune ore) e con una buona durata nel tempo (arco di ore).
Raggiunta la temperatura eutettica, nella quale le tre fasi (f = 3) sono in condizione di equilibrio, il sistema bicomponente (acqua e NaCl, c = 2) è invariante (v = c +1 -f = 2 +1 -3 = 0) e mantiene un valore costante di temperatura fino a quando non viene fuso tutto il ghiaccio o sciolto tutto il sale. Quando è verificata una di queste condizioni il sistema non è più zerovariante e quindi la sua temperatura tende nuovamente ad aumentare fino al raggiungimento della temperatura ambiente.
polverizzazione industriale
Per il processo di polverizzazione sono impiegati vari tipi di mulini, che producono polveri classificabili in base alla loro grandezza, come: grossolane (particelle > 850 mm); medie (paricelle comprese fra 850 e 75 mm) e fini (particelle < 75 mm).
Operativamente, i mulini possono lavorare a ciclo aperto o chiuso, a seconda che la riduzione delle particelle sia eseguita con o senza interruzioni. Durante questo processo il materiale è
sottoposto a forze che agiscono separatamente o congiuntamente:
- forze di impatto: agiscono perpendicolarmente alla superficie del solido in modo impulsivo;
- forze di attrito: agiscono tangenzialmente alla superficie del solido;
- forze di taglio: quando un solido è veicolato da un liquido i cui strati si muovono in una stessa direzione con velocità diverse, si trova sottoposto a più forze di taglio che lo sollecitano frantumandolo (su questo pricipio si basa il mulino colloidale);
- forze di pressione: sono forze simili a quelle di impatto, la cui durata è però prolungata nel tempo;
L'azione meccanica prodotta dalle forze citate, può essere insufficiente a determinare la rottura delle particelle: per esempio, nel caso di comportamento elastico del materiale con conseguente restituzione dell'energia applicata. Per questo, industrialmente si applicano forze in misura molto superiore al necessario, con conseguente dissipazione di energia termica e conseguente necessità di sistemi di raffreddamento. Ovviamente la durata del processo influenza il grado di finezza delle polveri, però occorre tener presente che quando le particelle raggiungono dimensioni dell'ordine di 10 mm, si può avere dissipazione di energia senza ottenere apprezzabili risultati sulla dimensione delle particelle, le quali, anzi, possono riaggregarsi.
Le caratteristiche specifiche dei vari mulini variano sensibilmente a seconda delle aziende produttrici; comunque, le principali tipologie possono essere riassunte nella tabella seguente.
| MULINO | AZIONE | DIMENSIONI POLVERI | ADATTO | INADATTO |
| coltello o lame | taglio | 850-200 | droghe vegetali e animali, grezze e fibrose | materiali frabili |
| sfere | attrito e impatto | 850-75 | materiale abrasivo | solidi soffici |
| martelli | impatto | 40-20 | quasi tutte | solidi soffici |
| cilindri | pressione | 850-75 | materiale soffice | materiale abrasivo |
| energia fluida | attrito e impatto | 30-1 | materiale poco friabile | materiale morbido e abrasivo |
| colloidale | taglio e vortici | 100-1 | materiale disperso in un fluido | materiali secchi |
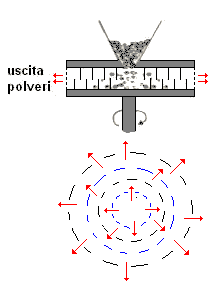
mulino a sfere
 è costituito da un recipiente cilindrico (v. fig. a dx)
contenenti sfere libere di muoversi; ambedue possono essere in metallo o porcellana. Ruotando il contenitore cilindrico, le sfere in esso contenute, seguiranno per attrito il suo movimento fino
ad una certa altezza, dalla quale per azione della forza di gravità ricadranno sul fondo, iniziando poi a saltare nel cilindro. E' importante che la forza centrifuga non raggiunga valori tali da
far aderire le sfere alla parete del cilindro, in quanto, in tal caso, si perderebbe l'azione dovuta alla compressione.
è costituito da un recipiente cilindrico (v. fig. a dx)
contenenti sfere libere di muoversi; ambedue possono essere in metallo o porcellana. Ruotando il contenitore cilindrico, le sfere in esso contenute, seguiranno per attrito il suo movimento fino
ad una certa altezza, dalla quale per azione della forza di gravità ricadranno sul fondo, iniziando poi a saltare nel cilindro. E' importante che la forza centrifuga non raggiunga valori tali da
far aderire le sfere alla parete del cilindro, in quanto, in tal caso, si perderebbe l'azione dovuta alla compressione.
|
m V2/R = m g |
dove:
D = diametro del cilindro;
V = velocità di rotazione del cilindro;
g = accelerazione di gravità
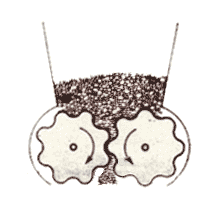
mulino a lame
 il prodotto da lavorare (v. fig. a dx) è sminuzzato passando tra
due piastre metalliche circolari e parallele, delle quali una è fissa e l'altra ruota ad alta velocità attorno al proprio asse.
il prodotto da lavorare (v. fig. a dx) è sminuzzato passando tra
due piastre metalliche circolari e parallele, delle quali una è fissa e l'altra ruota ad alta velocità attorno al proprio asse.
Le due piastre (a destra, parte in basso: le lame fisse sono colorate in blu) sono dotate di lame disposte concentricamente attorno all'asse di rotazione. L'uscita dal mulino ha una griglia
calibrata che lascia passare la polvere che si deposita in un cassetto di raccolta solo quando ha raggiunto le dimensioni richieste. Infatti, come mostra la formula nel riquadro, via via che le
dimensioni delle particelle si riducono, quelle di minore dimensione si allontanano dall'asse di rotazione:
|
F = m · v2/R = m · w2R |
dove:
R = raggio di curvatura delle particelle;
m = massa di una singola particella;
w = velocità angolare di rotazione del mulino
F = forza centripeta
mulino a cilindri

|
|
|
|
|
|
i cilindri di questi mulini possono essere scanalati o meno. La dimensione delle particelle è regolata dalla spaziatura dei due cilindri che combaciano reciprocamente durante la loro rotazione; le sostanze sono trascinate e schiacciate in questo spazio. Nel caso di due cilindri lisci, uno di questi ruota più velocemente dell'altro in modo da sommare un'azione di compressione e stiramento.
mulino a energia fluida
 Quando una particella di polvere ne colpisce un'altra con elevata velocità,
quest'ultima subisce una deformazione plastica. Quando la particella urtante rimbalza, alla deformazione si accompagnano fratture laterali, e la particella colpita è scheggiata (v. animazione a
sinistra). In una situazione pratica, centinaia di migliaia di particelle sono accelerate da un getto d'aria ad alta pressione e proiettate una contro l'altra alla velocità di 700 km/h.
Quando una particella di polvere ne colpisce un'altra con elevata velocità,
quest'ultima subisce una deformazione plastica. Quando la particella urtante rimbalza, alla deformazione si accompagnano fratture laterali, e la particella colpita è scheggiata (v. animazione a
sinistra). In una situazione pratica, centinaia di migliaia di particelle sono accelerate da un getto d'aria ad alta pressione e proiettate una contro l'altra alla velocità di 700 km/h.
Le macchine ad impatto, oltre che piuttosto ingombranti, richiedono un'enorme quantità di aria compressa, però sono molto efficienti e non provocano riscaldamento delle polveri; per questa
ragione sono molto diffuse nelle industrie farmaceutiche.
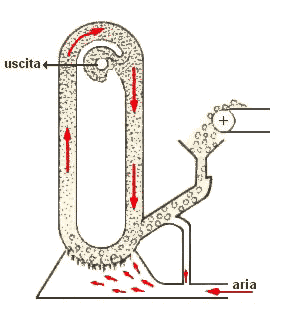 Nel mulino a energia fluida (v. fig. a dx), le
particelle da polverizzare vengono trascinate da una violenta corrente d'aria in una camera progettata in modo che subiscano un gran numero di urti reciproci.
Nel mulino a energia fluida (v. fig. a dx), le
particelle da polverizzare vengono trascinate da una violenta corrente d'aria in una camera progettata in modo che subiscano un gran numero di urti reciproci.
Via via che le dimensioni delle particelle si riducono, quelle di minore dimensione ruotano più vicine alla parete interna della camera:
|
F = m · v2/R |
dove:
R = raggio di curvatura delle particelle;
m = massa di una singola particella;
v = velocità delle particelle (dipende dal flusso d'aria e si può ritenere uguale per tutte);
F = forza centripeta
mulino a martello
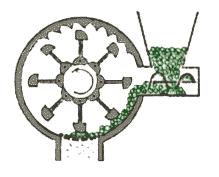 l'albero in rotazione (v. figura a destra) fa girare, ad alta
velocità, una serie di bracci metallici articolati, che polverizzano la sostanza da macinare provocandone l'urto contro le pareti del recipiente. L'uscita è calibrata in modo da permettere la
fuoriuscita delle polveri quando hanno raggiunto la dimensione richiesta.
l'albero in rotazione (v. figura a destra) fa girare, ad alta
velocità, una serie di bracci metallici articolati, che polverizzano la sostanza da macinare provocandone l'urto contro le pareti del recipiente. L'uscita è calibrata in modo da permettere la
fuoriuscita delle polveri quando hanno raggiunto la dimensione richiesta.
mulino colloidale:
(v. preparazione delle emulsioni)
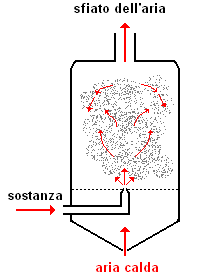
polverizzazione mediante nebulizzazione
Oltre ai processi meccanici esaminati, si può ricorrere alla polverizzazione mediante nebulizzazione. Ad es., si possono impiegare atomizzatori attraversati da un fluido costituito da una soluzione satura del solido da polverizzare, che viene spruzzata contro una corrente di aria calda: la rapida evaporazione del solvente lascia un residuo costituito da polveri finissime e soffici.
sospensioni
Le sospensioni sono sistemi termodinamicamente instabili, soggetti alla sedimentazione ed alla riaggregazione
delle particelle. Questa forma fisica rende possibile la somministrazione di solidi insolubili in formulazioni farmaceutiche più biodisponibili; si possono infatti allestire lozioni, unguenti,
preparazioni parenterali, orali e suppositorie.
 In particolare, per le preparazioni orali, possiamo distinguere fra
sospensioni già pronte e da preparare al momento dell'uso. Queste ultime sono confezionate, per motivi di stabilità, in modo che il solido sia fisicamente separato dal veicolo liquido (due fiale
separate, una con la polvere, l'altra con il solvente; oppure, tappo contenitore da premere al momento dell'uso, fig. a destra: sistema GePACK®); in questo caso, la data di scadenza riportata
sulla confezione, si riferisce al prodotto non ancóra ricostituito in quanto appena ottenuta la sospensione, la sua conservazione è garantita per periodi di tempo molto brevi.
In particolare, per le preparazioni orali, possiamo distinguere fra
sospensioni già pronte e da preparare al momento dell'uso. Queste ultime sono confezionate, per motivi di stabilità, in modo che il solido sia fisicamente separato dal veicolo liquido (due fiale
separate, una con la polvere, l'altra con il solvente; oppure, tappo contenitore da premere al momento dell'uso, fig. a destra: sistema GePACK®); in questo caso, la data di scadenza riportata
sulla confezione, si riferisce al prodotto non ancóra ricostituito in quanto appena ottenuta la sospensione, la sua conservazione è garantita per periodi di tempo molto brevi.
stabilità delle sospensioni
Per la preparazione delle sospensioni, è molto importante la determinazione delle dimensioni delle particelle
allo scopo di limitare la sedimentazione (per questa ragione le sospensioni devono sempre essere agitate al momento dell'uso).
In particolare, in accordo con il modello di Stokes, le particelle più grandi sedimentano più velocemente, ma sono facilmente ridisperdibili per agitazione; le particelle
più piccole sedimentano più lentamente, ma tendono a formare fondi impacchettati non più ridisperdibili (fenomeno detto "caking", incrostamento).
Per evitare questa eventualità, si ricorre a misure di potenziale zeta in modo da determinare la dimensione ottimale delle particelle disperse.
La suddivisione molto fine delle particelle può anche facilitare indesiderati fenomeni di adsorbimento; per questi motivi, eccezion fatta per le sospensioni da ricostituire prima dell'uso, si ricorre alla flocculazione. Questa tecnica consiste nel far sì che le particelle si aggreghino come flocculi, capaci di trattenere nel loro interno una parte di solvente. I flocculi precipitano più rapidamente, ma poiché costituiscono un sedimento abbastanza voluminoso sono facilmente ridisperdibili. D'altra parte, è importante che la velocità di sedimentazione dei flocculi sia tale da garantire una sospensione omogenea almeno per il tempo necessario al prelievo della dose da somministrare; ciò si può facilmente ottenere con opportuni agenti ispessenti, avendo cura che siano in quantità tali da permettere l'agitazione e lo scorrimento della sospensione.
Sebbene si possano ottenere sospensioni anche senza flocculazione, è utile definire il concetto di "grado di flocculazione", fg, definito dal seguente rapporto:
| fg = volume del solido sedimentato/volume del liquido = Vs/Vl |
Un ulteriore fattore che, per quanto possibile, si può prendere in considerazione, è la scelta di combinazioni solido-solvente con densità simile. In questo caso, come risulta dal modello di Stokes, la velocità di sedimentazione tende a zero.
preparazione industriale delle sospensioni
Il procedimento iniziale consiste nel ridurre il solido a particelle di adatte dimensioni, quindi le stesse vengono trattate con un agentebagnante e disperse nel mezzo; a questo punto, per ottenere dispersioni uniformi, si possono seguire tre criteri:
- le particelle non vengono flocculate ma incorporate in un liquido strutturato, cioé trattato con un agente ispessente che ne aumenta la viscosità e la densità. Per esempio, la gomma arabica presenta il fenomeno della tissotropia in quanto modifica la viscosità delle sospensione in misura variabile con l'intensità delle forze di taglio applicate (viscosità non Newtoniana); questo significa che il valore della viscosità può diminuire per agitazione, offrendo così il vantaggioo di stabilizzare la sospensione pur permettendo di poterla versare, prelevare in parte, iniettare, ecc.;
- le particelle vengono addizionate con un opportuno agente flocculante;
- le particelle vengono addizionate con un agente flocculante ed incorporate in un liquido strutturato. Poiché l'agente ispessente è quasi sempre un polianione, qualora si usi un flocculante carico positivamente, ad esempio Al, è chiaro che si avrà l'effetto negativo di una reciproca neutralizzazione. Per evitare questo inconveniente, si tratta inizialmente la sospensione, indipendentemente dallo strato elettrico dele particelle, con una sostanza (anche tensioattiva) dotata di una funzione amminica che circonderà le particelle con le sue cariche negative; a questo punto, si può aggiungere un agente flocculante, ad es., un fosfato.
Per la preparazione estemporanea di sospensioni, i problemi discussi non si pongono in quanto la semplice aggiunta di gomma arabica è sufficiente ad assicurare un'adeguata stabilità per il necessario periodo di tempo.
caratteristiche e proprietà delle emulsioni
Le emulsioni sono sistemi dispersi, cioè eterogenei, costituiti da due fasi liquide immiscibili tra loro. Possiamo distinguere emulsioni di olio in acqua (O/A, Oil/Water) ed emulsioni di acqua in olio (A/O, W/O). Nel primo tipo l'olio rappresenta la fase dispersa (o discontinua o interna) e l'acqua la fase disperdente (o continua o esterna); il contrario avviene per le emulsioni A/O. Per es., il latte ed il burro sono due emulsioni naturali, rispettivamente O/A e A/O.
emulsioni
Per stabilire il tipo di emulsione in esame, si può ricorrere a vari metodi:
- diluizione: se una data emulsione è diluibile in acqua (ad es., latte, maionese) è di tipo O/A; nel caso contrario è di tipo A/O (ad es., burro, margarina);
- uso di coloranti: un'emulsione addizionata con un colorante idrosolubile (ad es., blu di metilene), risulterà uniformemente colorata se è di tipo O/A. Se il colorante aggiunto all'emulsione è liposolubile (ad es., sudan - non per uso alimentare), si avrà colorazione uniforme se l'emulsione è di tipo A/O;
- conducibilità elettrica: poiché l'olio, al contrario dell'acqua conduce la corrente elettrica in misura estermamente ridotta, è ovvio che solo le emulsioni di tipo O/A permetteranno il passaggio di corrente fra due elettrodi;
- fluorescenza: la maggior parte deglio olii, e quindi le emulsioni A/O, emettono fluorescenza se eccitati con radiazioni elettromagnetiche di opportuna lunghezza d'onda.
Dal punto di vista farmaceutico, vengono utilizzate molte emulsioni (A/O e O/A) per uso topico, mentre è meno diffuso l'uso di emulsioni per uso orale (O/A); esistono infine anche particolari emulsioni multiple, ad es. O/A/O. Le dimensioni delle goccioline nella fase dispersa variano da 0.1 a 0.5 mm, ma esistono anche microemulsioni in cui le dimensioni scendono sotto il limite inferiore e ciò conferisce loro un aspetto trasparente in quanto non costituiscono più un ostacolo alla propagazione rettilinea delle luce (v.colloidi).

|
|
|
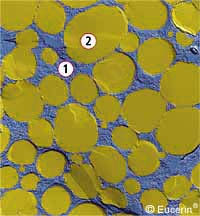
| immagine al microscopio elettronico di una emulsione O/A : 1) fase acquosa esterna; 2) fase oleosa interna (tratta dal sito: www.eucerin.co.uk) | immagine al microscopio elettronico di una emulsione O/A con maggior contenuto di olio: 1) fase acquosa esterna; 2) fase oleosa interna (tratta dal sito: www.eucerin.co.uk) |
Un problema fondamentale consiste nel prevedere il tipo di emulsione che si formerà dalla agitazione di due fasi immiscibili, e ciò dipende da vari fattori:
-
volume di fase: è il rapporto tra la percentuale della fase acquosa e quello della fase oleosa. L'entità di questo fattore è legata alla tendenza delle goccioline, che
costituiranno o dovranno costituire la fase dispersa, a coalescere, ovverosia a riunirsi tra loro per ricostituire una fase continua. In particolare, poiché la coalescenza aumenta in misura
proporzionale al numero di goccioline disperse, la fase presente in quantità maggiore avrà maggior tendenza a costituire la fase continua. D'altra parte, con opportuni tensioattivi e per
determinati valori di viscosità, si può limitare fino al 10% la percentuale di fase continua nell'emulsione; tuttavia, queste emulsioni sono poco stabili in quanto la fase presente in
quantità maggiore avrà sempre la tendenza a divenire la fase continua, con inversione dell'emulsione ad es. da O/A a A/O.
In pratica, è comunque raro che si preparino emulsioni in cui la fase continua sia percentualmente minore della fase discontinua, così, al massimo, in alcuni cosmetici si arriva ad un 60% di fase continua. -
viscosità delle fasi: la fase più viscosa si suddividerà meno facilmente producendo un minor numero di goccioline e quindi avrà maggior tendenza a costituire la fase
continua.
- tensioattivi: l'azione emulsionante di un tensioattivo si può spiegare facilmente pensando che costituisce una sorta di "ancoraggio" tra le gocciolone che costituiscono la fase discontinua ed il liquido che costituisce la fase continua. Per prevedere l'azione prodotta da un tensioattivo sulla dispersione di due liquidi immiscibili, si ricorre alla regola di Bancroft:la fase continua è quella in cui il tensioattivo è più solubile.
La regola di Bancroft, trova il suo fondamento teorico ove si consideri che l'abbassamento
della tensione superficiale di un liquido ne favorisce la frammentazione in gocce; sicché, se mescoliamo due liquidi immiscibili tra loro addizionandovi un tensioattivo, questo ridurrà in misura
minore la tensione superficiale del liquido in cui è più solubile (la riduzione della tensione avviene alla superficie di un liquido) e sarà quindi questo liquido che si comporterà da fase
esterna.
Così, ad es., poiché la solubilità in acqua di un tensioattivo aumenta con il valore di HLB, è logico che tensioattivi con elevato HLB diano prevalentemente emulsioni O/A.
Il fenomeno della coalescenza, fà sì che qualsiasi emulsione sia soggetta a destabilizzarsi; in questo processo possiamo distinguere tre stadi:
- flocculazione: le goccioline della fase discontinua iniziano a coalescere (questo stadio è reversibile);
- scrematura: il flocculato sale in superficie (stadio reversibile, ma più difficile da trattare);
- separazione delle fasi: l'emulsione si "rompe" definitivamente ed irreversibilmente.
Anche l'accennata inversione dell'emulsione può ritenersi, se indesiderata, destabilizzante:
in particolare, questa può verificarsi o quando il volume della fase discontinua è troppo elevato o per azione di un tensioattivo. Ad es., se si impiega come tensioattivo un sapone sodico
(idrosolubile) e nella fase continua sono presenti ioni calcio, questi trasformeranno il sapone in calcico (liposolubile) e ciò può favorire l'inversione.
stabilità delle emulsioni
Abbiamo discusso come individuare il tipo di emulsione in esame, come prevedere il tipo di emulsione che si formerà ed i suoi stadi destabilizzanti. Ora esamineremo i fattori che concorrono a stabilizzare le emulsioni.
-
agente emulsionante: generalmente è un tensioattivo, che abbassando la tensione interfacciale diminuisce l'energia libera del sistema; in alternativa, si possono
utilizzare anche sostanze non tensioattive, quali la gomma arabica, la gelatina, colloidi idrofili
o polveri finemente suddivise (ad es., talco). Queste sostanze si distribuiscono all'interfase O/A formando una pellicola protettiva, più rigida di quella formata dai tensioattivi, che
costituisce una barriera verso la coalescenza. Per contro, sostanze con forte affinità per l'acqua (come gli zuccheri) possono sottrarre acqua alle interfasi rompendo l'emulsione.
-
barriera all'interfase: la superficie definita dall'incontro tra la fase continua e le goccioline che costituiscono la fase discontinua, è elettrostaticamente carica
indipendentemente dalla natura ionica o non ionica del tensioattivo. Infatti, la carica sempre presente sulla barriera è dovuta o alla natura polare di una delle due fasi, o agli attriti (che
generano una carica elettrostatica), o ancóra all'adsorbimento di ioni presenti in soluzione (anche i tensioattivi si dispongono all'interfase per adsorbimento). E' quindi evidente che le
sostanze ionizzabili (come i sali) possono interferire con le cariche superficiali diminuendo le forze repulsive e facilitando la coalescenza.
- viscosità del mezzo, velocità di sedimentazione o scrematura, dimensione delle particelle: questi fattori sono correlati tra loro dalla legge di Stokes. Da questa legge si deduce che il sistema sarà tanto più stabile quanto più le densità delle due fasi sono vicine e quanto maggiore è la viscosità della fase continua; inoltre, si cercherà di ridurre quanto più possibile le dimensioni delle particelle, ottimizzandone le dimensioni (con successive prove sperimentali) in funzione del tensioattivo impiegato che ne controlla la stabilità.
scelta del tensioattivo
Per poter scegliere il tensioattivo più efficace, viene fatto corrispondere (v. tabella) alle più comuni fasi oleose, il cosiddetto HLBr(r = richiesto) del tensioattivo più adatto per formare l'emulsione più stabile.
| sostanza | HLBr O/A | sostanza | HLBr O/A |
| acido laurico | 16 | olio di oliva | 7 |
| acido linoleico | 16 | olio di ricino | 14 |
| acido oleico | 17 | olio di sesamo | 7 |
| acido stearico | 17 | olio di semi di carote | 6 |
| alcol cetilico | 15 | olio di semi di cotone | 7,5 |
| burro di cacao | 6 | olio di semi di girasole | 7 |
| cera carnauba | 12 | olio di soia | 7 |
| cera d'api | 9 | olio di vaselina | 10 |
| lanolina | 12 | paraffina | 10 |
| olio di avocado | 7 | olio di semi di mango | 7 |
| olio di cocco | 8 | olio di semi di borragine | 7 |
| olio di Jojoba | 6,5 | olio di arachidi | 6 |
Per determinare l'HLBr di un'emulsione, si preparano una decina di emulsioni stabilizzate da tensioattivi con HLB differenti, assegnando l'etichetta HLBr all'emulsione più stabile. Il valore dell'HLBr sarà ovviamente diverso a seconda che l'olio costituisca la fase continua o la fase discontinua. Noto il valore di HLBr, si può scegliere il tensioattivo più adatto, non solo come indice di HLB, ma anche in base a compatibilità chimiche e norme legislative. A tale scopo, è generalmente conveniente miscelare due tensioattivi, calcolando la loro presenza percentuale in miscela graficamente o analiticamente.
L'indice HLB è una grandezza estensiva (i suoi valori possono essere sommati algebricamente) e quindi è possibile prevedere gli effetti della combinazione risultante dall'associazione di due tensioattivi semplicemente calcolando la media aritmetica dei corrispondenti indici. Ad es., mescolando in parti uguali uno Span con HLB = 3 ed un Tween con HLB = 15, avremo una miscela con HLB = (3 + 15)/2 = 9. Questo fatto è molto utile in quanto per preparare certe emulsioni, sono richiesti dei particolari valori di HLBr che sono facilmente ottenibili miscelando due tensioattivi.
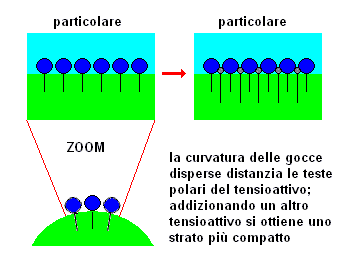 E' possibile
giustificare intuitivamente l'effetto della combinazione di due tensioattivi: si osservi la figura a destra, dove sono schematizzati due tipi di tensioattivi con affinità per le fasi acquosa ed
oleosa leggermente diversa. Risulta evidente la possibilità che i due tensioattivi si inseriscano nella fase acquosa in modo da formare uno strato più compatto.
E' possibile
giustificare intuitivamente l'effetto della combinazione di due tensioattivi: si osservi la figura a destra, dove sono schematizzati due tipi di tensioattivi con affinità per le fasi acquosa ed
oleosa leggermente diversa. Risulta evidente la possibilità che i due tensioattivi si inseriscano nella fase acquosa in modo da formare uno strato più compatto.
In generale, nella miscelazione di due tensioattivi, l'ottenimento di un particolare valore di HLB comporta che le concentrazioni dei due singoli tensioattivi siano diverse ed è possibile
valutarle sia analiticamente che graficamente. Supponiamo, per es., di voler determinare le percentuali, x ed y, di due tensioattivi caratterizzati
rispettivamente dai valori HLBx ed HLBy, necessarie per ottenere una miscela con indice HLBr.
Posto x + y = 1 (ossia la somma delle percentuali secondo cui vengono miscelati i due tensioattivi), risulta:
risolvendo rispetto ad x, ed esprimendo il risultato come percentuale, si ottiene:
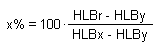
|
esempio: per concretizzare quanto discusso, si consideri la preparazione cosmetica riportata a destra, che essendo costituita da più componenti, richiede un calcolo a parte per l'HLBr:
| olio di vaselina | 33 parti |
| lanolina | 2 parti |
| alcol cetilico | 1 parte |
| emulsionante | 5 parti |
| acqua | q.b. a 100 |
Le singole percentuali dei componenti (escluso l'emulsionante), sono:
olio di vaselina = (33/36) · 100 = 91.66%
lanolina = (2/36) · 100 = 6.05%
alcol cetilico = (1/36) · 100 = 2.77%
poiché i rispettivi HLB riferiti alle sostanze singole sono: 10, 12, 15 (cfr. tabella precedente), il valore di HLBr richiesto per preparare l'emulsione più stabile con la loro associazione, sarà:
HLBr = (10 · 91.66 + 12 · 6.05 + 15 · 2.77 )/100 = 10.24
Per completare la preparazione, rimane da addizionare l'emulsionante (5 parti): per ottenere l'HLBr = 10.24 , si può miscelare uno Span con HLB = 3 e un Tween con HLB = 15
i due tensioattivi devono essere combinati nelle percentuali Tween 60% e Span 40% , che per 5 parti di emulsionante corrispondono a 3 parti di Tween e 2 parti di Span.
Il procedimento analitico può essere efficacemente sostituito utilizzando grafici già predisposti (come quello riportato in figura a destra), dove in ordinate sono riportati, da ambo il lati, i valori di HLB corrispondenti a vari tensioattivi, ed in ascissa una doppia scala fornisce i valori relativi delle concentrazioni espresse in percentuale. Il principio del metodo è basato sulla dipendenza della combinazione lineare dell'HLB dalle concentrazione.
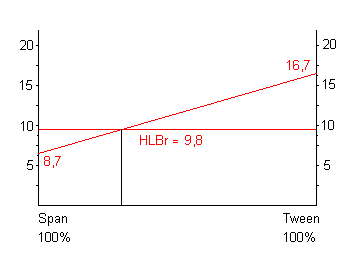 Supponiamo, ad es., di voler realizzare una miscela di tensioattivi per preparare un'emulsione il cui HLBr = 9.8, avendo a disposizione uno Span 40 (HLB = 6.7) ed un Tween 20 (HLBr = 16.7).
La miscelazione produce, in corrispondenza delle loro relative concentrazioni, una serie di valori HLB, riportati su una retta da 8.7 a 16.7, sicché la composizione percentuale della miscela
avente HLBr si ottiene dall'intersezione di questa retta con la retta parallela all'asse delle sacisse e passante per il valore HLBr = 9.8
Supponiamo, ad es., di voler realizzare una miscela di tensioattivi per preparare un'emulsione il cui HLBr = 9.8, avendo a disposizione uno Span 40 (HLB = 6.7) ed un Tween 20 (HLBr = 16.7).
La miscelazione produce, in corrispondenza delle loro relative concentrazioni, una serie di valori HLB, riportati su una retta da 8.7 a 16.7, sicché la composizione percentuale della miscela
avente HLBr si ottiene dall'intersezione di questa retta con la retta parallela all'asse delle sacisse e passante per il valore HLBr = 9.8
Il calcolo numerico può essere impiegato per la determinazione dell'HLB di un tensioattivo. Si procede in questo modo: si preparano alcune miscele, in varie proporzioni, di un tensioattivo avente HLBn (n = noto) con il tensioattivo di cui non si conosce l'HLBi (i = incognito). Poi, con queste miscele, si prepara una emulsione di cui si conosca l'HLBr. Alla miscela di tensioattivi che dà l'emulsione più stabile, si assegna il valore di HLBr e quindi si calcola l'HLBi con questo procedimento:
posto x + y = 1 (ossia la somma delle percentuali secondo cui vengono miscelati i due tensioattivi), risulta:
essendo noti HLBn ed HLBr, risolvendo questa equazione, si ricava HLBi
due classiche emulsioni galeniche
L'emulsione riportata nella ricetta in basso a sinistra, è una classica emulsione lassativa, la cui azione
deriva dalla presenza dell'olio di vaselina. In questo caso, per mascherarne il sapore, si formula un'emulsione O/A edulcorata con saccarosio.
La stabilizzazione di questa emulsione, che si presenta come una forma sciropposa piuttosto viscosa, non è
affidata alla presenza di tensioattivi, bensì alle gomme, che ne aumentano la viscosità ostacolando la separazione delle fasi.
L'emulsione riportata nella ricetta in basso a destra, è la cosiddetta cold cream. In questo
caso, per conferire caratteristiche più adatte all'uso topico, si formula un'emulsione A/O .
La stabilizzazione di questa emulsione, che si presenta come una forma cremosa, è affidata alla presenza di un tensioattivo prodottoin situ dalla salificazione dell'acido cerotico -
contenuto nella cera - con l'acido borico.
Le due preparazioni citate sono riportate nella loro formulazione classica e quindi il tipo e la quantità di tensioattivo non deve essere calcolata (come è stato fatto negli esempi proposti): è
un pò come la pasta alla carbonara, si può modificarne la ricetta (come faccio io perché sono vegetariano) ma allora non è più pasta alla carbonara... è un'altra cosa.
|
metodi per la preparazione delle emulsioni
Quanto discusso a proposito delle proprietà generali delle emulsioni, ci mette in grado di esaminare i metodi per la loro preparazione, galeniche ed industriali:
-
metodo continentale: noto anche come della gomma secca o della gomma nella fase interna, è caratterizzato da proporzioni sempre fisse, 1:2:4, tra agente emulsionante,
fase acquosa e fase oleosa. Questo metodo, consiste nel disperdere una parte di agente emulsionante (quasi sempre gomma arabica) in quattro
parti di fase oleosa, ottenendo una massa omogenea alla quale vanno aggiunte lentamente due parti di acqua. Data l'iniziale prevalenza della fase
oleosa, si forma prima una emulsione A/O, che poi, con la successiva aggiunta dell'acqua, si inverte formando un'emulsione O/A che può essere ulteriormente lavorata con acqua.
-
metodo inglese: noto anche come della mucillagine, della gomma nella fase esterna o della maionese, consiste nel preparare una mucillagine di gomma arabica in acqua; a
questa si aggiunge un pò di fase oleosa e si tritura finché la miscela raggiunge una fluidità costante. Si aggiunge quindi altra acqua e poi altro olio, fino ad esaurire entrambi. In
generale, qualora occorra sciogliere una sostanza (idro o liposolubile) nell'emulsione, conviene farlo prima che questa si formi, per evitare possibili rotture.
- metodo della bottiglia: è usato quando la componente oleosa è volatile. In pratica si mettono tutti i componenti in una bottiglia, poi la si chiude e si agita vigorosamente fino a formare l'emulsione. Questo metodo riflette la tecnica delle preparazioni industriali.
Nella preparazione industriale, tutti i metodi esaminati vengono eseguiti in grandi recipienti muniti di agitatori meccanici; le emulsioni così ottenute possono poi essere raffinate con molini colloidali ed omogenizzatori, che riducono ulteriormente le dimensioni delle particelle disperse.
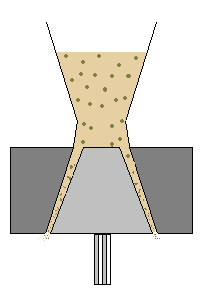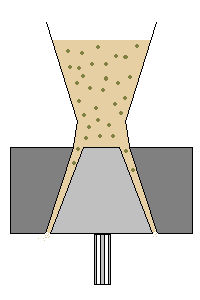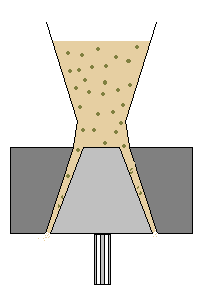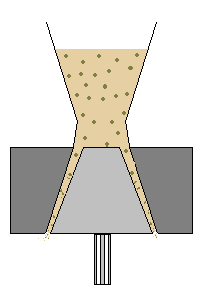
Il molino più diffuso è quello colloidale (figura a sinistra), costituito da un rotore tronco-conico che ruota, ad altissima velocità, internamente ad uno statore. La distanza fra rotore e statore può essere regolata in modo da ottenere la desiderata consistenza dell'emulsione. L'omogenizzatore (figura a destra) è costituito da una filiera coassiale e da un ugello. L'emulsione grossolana viene laminata ed omogenizzata attraversando l'ugello la cui sezione può essere variata agendo su una vite micrometrica collegata alla filiera. L'emulsione viene lavorata più volte e riversata nuovamente nella miscela fino ad ottenere la desiderata omogeneizzazione.
Il molino colloidale MK 2000 (www.ikausa.com/mk.htm) è specialmente progettato per la produzione di miscele colloidali, emulsioni estremamente fini e sospensioni. L'alta velocità di punta, combinata con scanalature (shear gap) estremamente ridotte produce un'intensa frizione sul materiale trattato. La frizione ed il taglio che si producono sono comunemente note come molatura umida. Il rotore e lo statore sono a forma conica ed hanno tre fasi delle dentellature sempre più fini. Lo statore può essere registrato micrometricamente per ottenere la necessaria distanza che fra il rotore e lo statore. Le scanalature cambiano i versi in ogni fase per aumentare la turbolenza. Con i rivestimenti ed i materiali di alta qualità, l'apparecchiatura Mk offre una geometria di macinazione estremamente efficiente.

|

|
controlli delle emulsioni farmaceutiche
Queste
preparazioni sono sottoposte a varie condizioni drastiche: prove gravitazionali, in cui l'emulsione da controllare viene centrifugata per analizzare successivamente le dimensioni delle
particelle; prove di escursioni termiche, congelando e scaldando l'emulsione ed osservandone il comportamento.
I dati ottenuti da queste ed altre prove sono poi confrontati con quelli standard relativi ad un'emulsione
campione. Infine, c'è il controllo reologico in cui si verifica, ad es., il comportamento
della viscosità in corrispondenza all'applicazione di forze di taglio, oppure, nel caso di preparazioni dermatologiche, il comportamento della spandibilità. Altri esami possono essere
semplicemente quelli di invecchiamento, lasciando il prodotto in adatti locali e controllandone le caratteristiche di stabilità.
liofilizzazione
La liofilizzazione è un processo tecnologico che consente l'eliminazione totale dell'acqua degli alimenti (e
farmaci), i quali vengono ridotti in polveri disidratate che per aggiunta della giusta quantità di acqua assumono il gusto e le caratteristiche nutritive (o terapeutiche) che avevano i prodotti
prima del trattamento.
Per comprendere i princìpi base di questo processo, occorre far riferimento al diagramma di stato
dell'acqua.
diagramma di stato dell'acqua
Lo studio dei cambiamenti di fase dell'acqua è essenziale per comprendere il processo di liofilizzazione. I cambiamenti di fase sono visualizzati tramite il diagramma di stato (o di fase) dell'acqua.
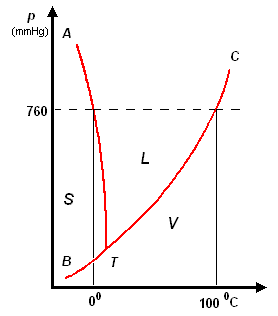 Il diagramma a destra (in scala arbitraria per
evidenziare la curvatura di TA verso temperature basse) rappresenta gli stati fisici in cui si trova l'acqua al variare di pressione e temperatura.
Il diagramma a destra (in scala arbitraria per
evidenziare la curvatura di TA verso temperature basse) rappresenta gli stati fisici in cui si trova l'acqua al variare di pressione e temperatura.
si distinguono tre zone:
- S = zona di esistenza della fase solida
- L = zona di esistenza della fase liquida
- V = zona di esistenza della fase vapore
e tre curve a delimitazione delle zone di cui sopra:
- BT = curva di coesistenza delle fasi solido-vapore; corrisponde ai processo di sublimazione-brinamento
- TC = curva di coesistenza delle fasi liquido-vapore; corrisponde ai processi di evaporazione-condensazione
- TA = curva di coesistenza delle fasi solido-liquido; corrisponde ai processi di fusione-solidificazione
Infine, l'intersezione in T delle tre curve, rappresenta il punto triplo di coesistenza delle fasi solido-liquido-vapore.
Il punto triplo T è caratterizzato da una unica coppia di valori per p e per t: 4,58 mm Hg e 0,01°C. Questo punto non coincide con
il punto di fusione (p = 760 mmHg; t = 0,00°C), poiché questa avviene in presenza di una pressione esterna, p, esercitata dall'aria, come
evidenziato nel diagramma.
La curva TA rappresenta così la variazione del punto di fusione sotto l'effetto di una pressione, p, esterna. Ovviamente, sopra al punto critico (temperatura critica 374°C, pressione critica 218 atm) non può esistere equilibrio liquido-vapore L-V, poiché l'acqua esiste solo allo stato vapore.
 L'inclinazione di TA verso sinistra,
al crescere della pressione, è una caratteristica dell'acqua, e dà ragione del fatto che aumentando la pressione, p, sul ghiaccio a 0°C, questo passa da fase solida a
liquida. Questo fenomeno è sfruttato, per esempio, nel pattinaggio sul ghiaccio. Infatti, la pressione esercitata dalle lame del pattino provocano una fusione superficiale del ghiaccio: il velo
d'acqua liquida che si produce, facilita lo scorrimento della lama sul ghiaccio; quando la pressione torna al livello normale, il velo di acqua solidifica nuovamente.
L'inclinazione di TA verso sinistra,
al crescere della pressione, è una caratteristica dell'acqua, e dà ragione del fatto che aumentando la pressione, p, sul ghiaccio a 0°C, questo passa da fase solida a
liquida. Questo fenomeno è sfruttato, per esempio, nel pattinaggio sul ghiaccio. Infatti, la pressione esercitata dalle lame del pattino provocano una fusione superficiale del ghiaccio: il velo
d'acqua liquida che si produce, facilita lo scorrimento della lama sul ghiaccio; quando la pressione torna al livello normale, il velo di acqua solidifica nuovamente.
Il diagramma di stato, è tracciato mediante l'equazione di Clausius-Clapeyron:
|
|
dove:
dp/dT = variazione della pressione con la temperatura;
DH = variazione di entalpia associata alla transizione di fase;
T = temperatura assoluta;
DV = variazione di volume associata alla transizione di fase.
il rapporto che compare a primo membro dell'eq. di Clausius-Clapeyron permette di valutare la variazione della pressione con la temperatura. La combinazione delle grandezze a secondo membro, può dare un risultato positivo o negativo, in corrispondenza dei valori assunti da DH e DV (la temperatura assoluta, T, è sempre positiva per definizione). Nel caso dell'acqua, si ha:
-
transizione solido
 liquido: il processo richiede
calore, quindi DH > 0, contemporaneamente si ha una diminuzione di volume DV < 0;
liquido: il processo richiede
calore, quindi DH > 0, contemporaneamente si ha una diminuzione di volume DV < 0;
-
transizione solido vapore: il processo richiede
calore, quindi DH > 0, contemporaneamente si ha un aumento di volume DV > 0;
-
transizione liquido vapore: il processo richiede
calore, quindi DH > 0, contemporaneamente si ha un aumento di volume DV > 0;
E' facile verificare che solo nella prima transizione di fase il
termine a secondo membro è negativo e quindi, risultando dp/dT < 0, la pressione di vapore del sistema solido-liquido diminuisce con l'aumentare della temperatura. Questo è
l'unico tratto del diagramma di stato dell'acqua con pendenza negativa (caratteristica anche di antimonio, bismuto e gallio) che presenta una diversa inclinazione delle superfici di separazione
solido - liquido. Tali sostanze, infatti, durante il processo di solidificazione aumentano il volume specifico.
Per integrazione dell'equazione di Clausius-Clapeyron, si ottiene la formula:
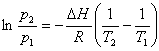
|
che permette di calcolare (per un certo valore di DH), nota la pressione di vapore ad una data temperatura, la pressione di vapore ad una qualsiasi temperatura.
La pressione di vapore (o tensione di vapore) di un liquido aumenta con la temperatura in misura data dall'equazione di Clausius-Clapeyron. Questa relazione assume che
si formi un gas ideale via via che un liquido evapora, ed è valida per molte sostanze.
Per i liquidi esiste una temperatura critica oltre la quale lo stato liquido non è stabile. In altre parole, quando una sostanza è riscaldata oltre la temperatura
critica, esiste come un fluido: liquido e vapore diventano indistiguibili.
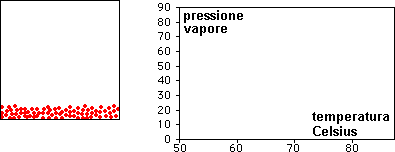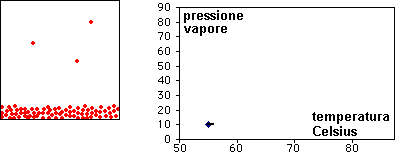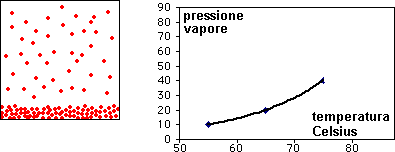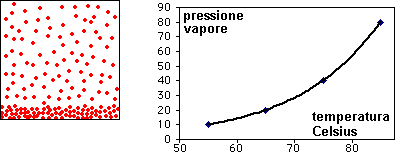 l'animazione mostra l'aumento della pressione della fase gassosa con la temperatura al di sotto della temperatura critica. |
il processo di liofilizzazione
La liofilizzazione avviene a bassissime temperature ed in condizioni di vuoto spinto in modo che l'acqua
contenuta nel prodotto previamente congelato si trasformi in ghiaccio e sublimi passando dallo stato solido a quello di vapore.
La liofilizzazione può definirsi come un processo di disidratazione controllata di prodotti preventivamente
congelati che consente di ottenere, oltre a farmaci in polveri liofilizzate ed alimenti conservati, con caratteristiche molto simili a quelle dei prodotti originali, diversi vantaggi,
quali:
- lungo periodo di conservazione (superiore ai due anni) anche a temperatura ambiente, se adeguatamente confezionato;
- protezione dagli inquinamenti da parte dei microorganismi;
- ineccepibilità igienica;
- facilità di trasporto e magazzinaggio del prodotto;
- rapida ricostituzione del prodotto non appena il liofilizzato viene posto in presenza dell'acqua;
- conservazione delle caratteristiche del prodotto di partenza (il patrimonio proteico, il contenuto in vitamine, gli elementi minerali ed i lipidi, la cui conservazione serve a garantire la buona digeribilità del prodotto).
Si tratta, in pratica, di congelare il prodotto da trattare ad una temperatura di -30, -40 °C all'interno di recipienti in acciaio inox (rispondenti alle norme igienico sanitarie), i quali, a loro volta, vengono posti all'interno del liostato in cui la pressione viene ridotta ad un valore tale che l'acqua presente nel prodotto precedentemente congelato, possa sublimare sotto vuoto mediante il riscaldamento ad una temperatura di 30° C, lasciando il prodotto essiccato praticamente in maniera completa.
Si ottiene così una massa solida, porosa, friabile, igroscopica, molto solubile nel medesimo solvente, che occupa lo stesso volume della massa congelata iniziale, detta liofilizzato. La ragione principale dello sviluppo di questa tecnologia nel settore farmaceutico è dovuta alla possibilità di realizzare formulazioni stabili nel tempo utilizzando sostanze che si degradano facilmente in soluzione.
Liofilizzato è, per esempio, tutto il cibo consumato sino ad oggi dagli astronauti nello spazio. I liofilizzati conservano le stesse qualità nutrizionali dei prodotti di partenza e vanno reidratati prima del consumo: si tratta di un'operazione pressocché istantanea e l'alimento ricostituito è del tutto simile a quello fresco.
operazioni relative al processo di liofilizzazione
1) fase preparatoria: comprende le operazioni di dissoluzione o sospensione nel solvente delle sostanze attive e dell'eventuale supporto; filtrazione della soluzione (quando necessario con effetto sterilizzante seguìta da immediato trasferimento del filtrato in blocco sterile), ripartizione nei contenitori costituiti generalmente da fiale o flaconi.
Poiché molto spesso la sostanza attiva per ogni contenitore è in quantità troppo piccola (anche pochi mg) ed il volume di soluzione corrispondente sarebbe difficilmente dosabile con precisione, oltre al fatto che alla fine del processo la fiala o il flacone sembrerebbero quasi vuoti, è necessario aggiungere un eccipiente, che sia atossico, privo di qualsiasi attività farmacologica e dia un liofilizzato di bell'aspetto: uno degli eccipienti utilizzati, é il mannitolo.
 rappresentazione schematica di un'apparecchiatura per liofilizzazione (la figura non è in scala e i dettagli sono incompleti) |
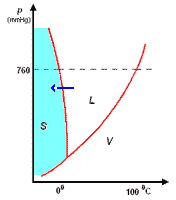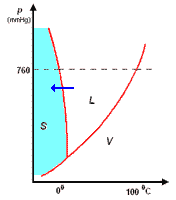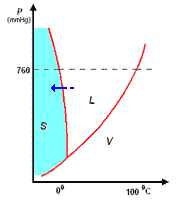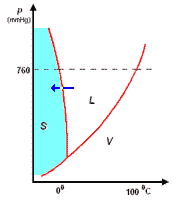 La soluzione o la sospensione viene congelata mediante raffreddamento rapido |
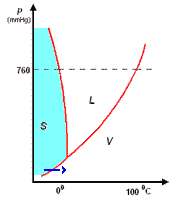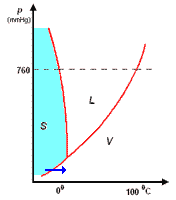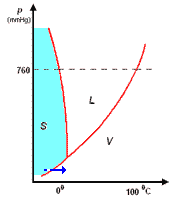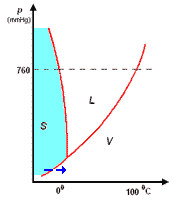 Il solvente rimasto nel liofilizzato viene eliminato mediante sublimazione |
2) fase di liofilizzazione: comprende:
a) il congelamento della soluzione ripartita, può essere condotto velocemente o lentamente, però generalmente lo si esegue in modo rapido perché in tal modo la formazione di cristalli di ghiaccio si produce simultaneamente in tutte le parti del preparato e si ottengono cristalli piccoli e distribuiti uniformemente: la formazione di strutture cristalline di grandi dimensioni derivanti da un processo di congelamento lento può danneggiare il prodotto da liofilizzare (aspetto particolarmente critico per la struttura cellulare di materiali organici). Nella liofilizzazione industriale il congelamento avviene per raffreddamento. Il materiale può essere congelato nella stessa camera in cui avviene l'essiccamento o al di fuori, in contenitori detti precongelatori o frigocelle, ponendolo su piastre portaprodotto che vengono raffreddate. E' opportuno disporre il materiale su ampia superficie e con spessore ridotto al minimo per facilitare le fasi successive di liofilizzazione.
b) la sublimazione del ghiaccio (o essiccamento), che a sua volta prevede due punti:
-
essiccamento primario: una volta congelato omogeneamente in tutte le sue parti, il preparato viene sottoposto all'azione del vuoto per provocarne la sublimazione del
solvente. Quando il vuoto raggiunge valori elevati si inizia il riscaldamento. Durante la sublimazione è necessario che il materiale sia perfettamente congelato, in caso contrario si possono
verificare fenomeni di fusione della massa con possibilità di alterazione del prodotto. Per questa ragione è necessario conoscere il valore della temperatura alla quale la massa congelata
comincia a fondere, onde poter fissare la zona di lavoro più idonea. Sono necessarie due condizioni per avere una continuità nel processo: un continuo apporto di calore alle molecole del
congelato e l'evacuazione progressiva del vapore prodotto.
Durante il processo di sublimazione, è necessario fornire calore al prodotto; diversamente, la sua temperatura diminuirebbe fino ad arrestare il processo di essiccamento. Per questa ragione si utilizza un termostato che riscalda i ripiani in modo che possano essere usati per fornire il calore necessario a rimpiazzare l'energia persa con l'evaporazione dell'acqua di sublimazione in modo da mantenere il prodotto a temperatura costante.
L'eliminazione del vapore acqueo (necessaria per impedire che la camera di sublimazione si saturi di vapore acqueo) viene effettuata mediante condensatori (apparecchiature fornite di superfici refrigerate sulle quali il vapore si deposita ricondensandosi direttamente come ghiaccio). La temperatura del condensatore è di circa 20°C inferiore a quella della massa in corso di liofilizzazione, per permettere il passaggio del vapore dalla camera di sublimazione a quella di condensazione in base alla differenza di tensione di vapore del ghiaccio esistente in queste due zone e tenuto conto che in entrambe le camere sono mantenuti gli stessi valori di depressione. -
essiccamento secondario: quando l'ultimo frammento di ghiaccio è scomparso, si può ritenere terminato l'essiccamento primario: ciò si può apprezzare praticamente
attraverso il rialzo di temperatura della massa. L'essiccamento secondario è detto anche desorbimento perché si riferisce all'eliminazione del solvente adsorbito tramite un ulteriore
innalzamento di temperatura. L'essiccamento secondario serve a ridurre l'umidità dal 7 all'1% circa del peso secco e ad aumentare la conservabilità del prodotto essiccato.
3) fase conclusiva: comprende l'esclusione del vapore acqueo dal sistema mediante ricongelamento e raccolta su di un apposito condensatore, l'apertura della camera di liofilizzazione e la chiusura delle fiale o dei flaconi.
 Il processo di liofilizzazione - mentre il
materiale essicca gradualmente - richiede molte ore (anche giorni - nell'animazione a sinistra è schematizzato un processo che dura 8 ore, accelerato 5.000 volte) in quanto l'eccessivo
riscaldamento del materiale (per affrettare il processo) può produrre significativi cambiamenti di composizione e struttura. Inoltre, l'accelerazione del processo di sublimazione potrebbe
produrre vapor d'acqua in quantità maggiore di quella che viene rimossa per azione della pompa da vuoto. Questo potrebbe parzialmente reidratare il materiale degradandone la qualità.
Il processo di liofilizzazione - mentre il
materiale essicca gradualmente - richiede molte ore (anche giorni - nell'animazione a sinistra è schematizzato un processo che dura 8 ore, accelerato 5.000 volte) in quanto l'eccessivo
riscaldamento del materiale (per affrettare il processo) può produrre significativi cambiamenti di composizione e struttura. Inoltre, l'accelerazione del processo di sublimazione potrebbe
produrre vapor d'acqua in quantità maggiore di quella che viene rimossa per azione della pompa da vuoto. Questo potrebbe parzialmente reidratare il materiale degradandone la qualità.
-
fine liofilizzazione: per verificare la fine della liofilizzazione si eseguono due o tre prove di perdita vuoto nella camera dove è posto il prodotto isolandola dal
condensatore tramite la chiusura di una valvola intermedia. Se la perdita di vuoto è contenuta e costante si ritiene finita la liofilizzazione.
-
chiusura dei contenitori: i prodotti liofilizzati sono altamente igroscopici, quindi è necessario che vengano chiusi in idonei contenitori a tenuta, operando in locali
per lo più sterili con umidità relativa molto bassa.
I cibi liofilizzati vengono confezionati, in atmosfera protetta o controllata, con involucri resistenti all'ossigeno e all'umidità, generalmente alluminio e polietilene, ma anche vetro. Si applica a: caffè, the solubile, camomilla solubile, succhi di frutta, frutta esotica, funghi prodotti dietetici e per l'infanzia, farmaci. Quanto più il confezionamento è accurato, tanto più il prodotto può mantenersi integro per anni e anni senza degradarsi finquando è riportato alla sua forma originale con un po' d'acqua.
Le fiale vengono saldate alla fiamma con apposite macchine automatiche. I flaconi vengono inseriti in una macchina che li chiude con il tappo perforabile con l'ago della siringa, in gomma butile (il materiale più idoneo per la sua alta impermeabilità ai vapori d'acqua), inserisce la capsula di alluminio e la ghiera sul collo del flacone. Nella parte centrale, superiormente, la capsula ha uno sportellino a disco, sollevabile al momento dell'uso, per l'introduzione dell'ago.
vantaggi della liofilizzazione
L'uso della liofilizzazione si impone qualora la sostanza da essiccare si alteri nel corso dell'essiccamento con altre metodiche. In tal caso, infatti, le basse temperature utilizzate nella liofilizzazione impediscono cambiamenti chimici che possono verificarsi a carico di numerosi componenti termolabili presenti in farmaci, alimenti, sostanze naturali. La liofilizzazione impedisce anche la formazione di schiuma o bolle in quanto l'essiccamento avviene allo stato solido; ciò è particolarmente importante per le soluzioni proteiche, in cui la formazione di schiuma può comportare denaturazione delle proteine. Inoltre, la liofilizzazione assicura la dispersione permanente del materiale, che si essicca senza subire concentrazione, e forma un solido poroso che si scioglie all'occorrenza con grande rapidità ed efficacia. Un ulteriore vantaggio è che in corso di liofilizzazione le particelle di soluto restano praticamente bloccate nelle posizioni che occupavano prima dell'evaporazione del solvente, evitando quindi effetti di coagulazione. Importantissimo è poi il fatto che la crescita batterica ed i cambiamenti enzimatici non possono aver luogo nel materiale congelato durante liofilizzazione ed i liofili ottenuti sono molto resistenti a tali indesiderabili fenomeni.
granulati
la granulazione è un processo che permette di trasformare polveri cristalline o amorfe in aggregati solidi che presentano caratteristiche più favorevoli per le lavorazioni industriali.
- scorrimento: i granulati hanno forma più regolare ed omogenea rispetto alle polveri di partenza;
- dosabilità: poiché i sistemi di misura meccanizzati sono sempre volumetrici, ad una maggior omogeneità di dimensioni corrisponderà una maggior uniformità di volume ed una conseguente maggior uniformità nel riempimento e nel peso delle singole dosi forma, esigenza particolarmente rilevante nel caso di farmaci ad elevata attività;
- comprimibilità: per la produzione di compresse, i granulati sono più adatti delle polveri;
- ripartizione: le polveri di partenza possono avere densità diverse e dato che le macchine impiegate nei processi di lavorazione producono spesso vibrazioni, si può avere la segregazione (le particelle più piccole scivolano attraverso i vuoti tra le particelle più grandi) di un componente della miscela: si potrebbe determinare, per es. nella fase di ripartizione volumetrica delle compresse, l'arricchimento di un componente sul loro fondo, con conseguente composizione non uniforme. I granuli, data la loro maggiore omogeneità, non presentano questo inconveniente.
preparazione dei granulati
Il processo di granulazione a letto fluido (noto anche come agglomerazione), coinvolge particelle sospese in un flusso d'aria ed un liquido spruzzato dall'alto verso il basso all'interno del letto fluido. Le particelle immerse nella traiettoria dello spray divengono leggermente bagnate e quindi appiccicose. Le particelle appiccicaticce urtano con altre perticelle ed aderiscono a queste formando un granulo.
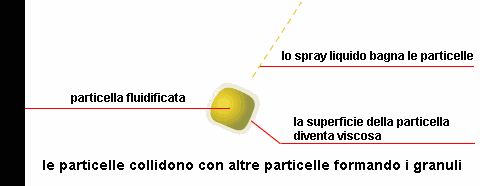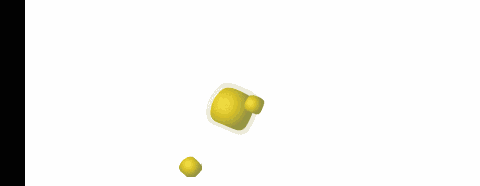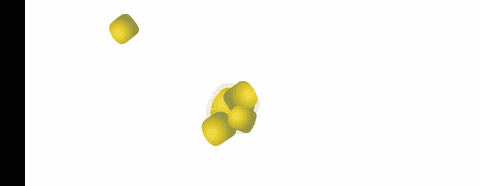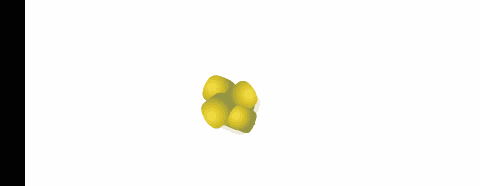

Vi sono due differenti modalità di granulazione a letto fluido: in fase secca o bagnata. Nella granulazione in fase secca, le particelle richiedono solo una leggera bagnatura per diventare
collose e riunirsi l'una con l'altra. La soluzione granulante è applicata ad una velocità minore o uguale alla velocità di evaporazione, in modo che le particelle rimangano secche durante
l'intero processo.
Nella granulazione in fase umida, le particelle richiedono un maggior cospargimento prima che diventino abbastanza collose da potersi riunire l'una con l'altra. La soluzione granulante è
applicata ad una velocità maggiore di quella di evaporazione finché le particelle raggiungono l'umidità sufficiente per la granulazione. Le caratteristiche delle particelle bagnate ed il tipo di
soluzione granulante usato, determinano quale debba essere il procedimento più appropriato. Mentre la fase secca è più comune, la fase umida permette di ottenere prodotti pù densi.
L'animazione, parzialmente convertita da Flash e adattata in gif animata, è stata tratta dal sito: www.vectorcorporation.com/flash/fluidbed.htm
Copyright ©2000 Fluid Air Incorporated. All rights reserved
granulati effervescenti
I granulati possono anche essere utilizzati come preparazioni farmaceutiche multidose, spesso rese effervescenti per migliorarne la palatabilità, come per esempio per la Citrosodina®. In questo caso, oltre ad impiegare recipienti ben chiudibili, occorre minimizzare la presenza di acqua per evitare che l'effervescenza prodotta dall'anidride carbonica (liberata dall'acido tartarico o citrico) si sviluppi durante la preparazione del granulato.
I granulati effervescenti contengono componenti a reazione acida e carbonati o bicarbonati in grado di reagire rapidamente, in presenza di acqua, liberando anidride carbonica.
Un comune metodo di preparazione prevede bicarbonato di sodio e acido citrico (di cui il 20-25% monoidrato), unitamente ad altri componenti miscelati e riscaldati fino alla liberazione di acqua di cristallizzazione dell'acido citrico che agisce da agente granulante. In questo caso il contenuto riscaldamento e la limitata quantità di acqua provocano una modesta liberazione di anidride carbonica che contribuisce a rendere spugnosa la massa. Come dolcificante, si addizionano quelli previsti dalla legge.
Se per esempio si vogliono preparare 10 bustine da 5 g ciascuna in granulato effervescente contenente 500 mg di acido acetilsalicilico per bustina, le relative quantità dei componenti si calcolano come segue:

La miscela effervescente (45 g) è costituita da: acido citrico, acido tartarico (acidificante, esaltatore di sapidità), bicarbonato di sodio
Il calcolo delle quantità di acido citrico e bicarbonato da utilizzare è basato sull'equazione stechiometrica:
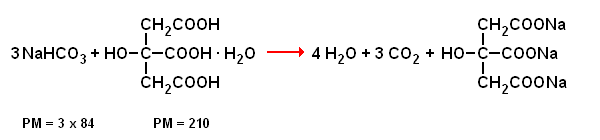
Dalla stechiometria della reazione acido citrico + bicarbonato, risulta che per 1 g di acido citrico sono necessari 1,2 g di bicarbonato (1 : 210 = x : 252).
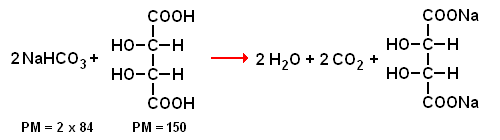
Dalla stechiometria della reazione acido tartarico + bicarbonato, risulta che per 1 g di acido tartarico sono necessari 1,12 g di bicarbonato (1 : 150 = x : 168)
| bicarbonato | ac. citrico | ac. tartarico |
| 1.2 | 1 | |
| 1.12 | 1 | |
|
g di bicarbonato necessari rispettivamente per 1 g di ac. citrico e 1 g di ac. tartarico |
||
Comunemente l'ac. citrico e l'ac. tartarico vengono addizionati nel rapporto 1:2 ; pertanto, miscelando gli acidi secondo questa proporzione, occorrono:
1 g ac. citrico + 1,2 g di bicarbonato + 2 ·[1 g ac. tartarico + 1,12 g di bicarbonato] = 6,4 g di miscela effervescente.
Ora, occorre calcolare le quantità di miscela effervescente necessarie per i 45 g richiesti per ogni bustina: occorrono 45/6,4 = 7 unità di miscela (acido citrico, ac. tartarico, bicarbonato). In
pratica, rispettando i rapporti dei componenti abbiamo: 7 ac. citrico + 14 ac. tartarico + 24 [ = 7 · (1,2 + 2,22)] sodio bicarbonato = 45 g. A questa miscela si addizioneranno i 5 g di acido
acetilsalicilico.
Uno dei problemi nella produzione dei granulati effervescenti è costituito dalla presenza di umidità sia durante la preparazione sia nel prodotto finito. Il prodotto viene protetto introducendo nel tappo del contenitore un essiccante (gel di silice, solfato di calcio anidro).
La formulazione esaminata è stechiometrica; tuttavia, per variare il gusto, e per raggiungere il volume desiderato è possibile miscelare i componenti in quantità non stechiometrica. Per
es., ciascuna tavoletta di Alka-Seltzer® contiene acido acetilsalicilico 324 mg, sodio bicarbonato 1625 mg, acido citrico anidro 965 mg
L'acido citrico da solo è sufficiente per conferire un carattere frizzante all'acqua, unitamente al gusto (aspro) di limonata. Invece, addizionando l'acido tartarico - che produce sulla lingua e
sul palato una sensazione di fresco accompagnata dal sapore salato del tartrato (ottenuto dalla salificazione dell'ac. tartarico) - all'acido citrico (sapore aspro), c'è un sinergismo tra i due
sapori.
| sapore di riferimento | dolce | amaro | salato | aspro |
| dolce | attenuato | equilibrato | attenuato | |
| amaro | equilibrato | rinforzato | rinforzato | |
| salato | attenuato | esaltato | esaltato | |
| aspro | attenuato | rinforzato | rinforzato |
La tabella a fianco aiuta a comprendere la relazione fra i vari sapori e le differentti sensazioni derivanti dalle loro combinazioni. Partendo dalla colonna contenente i sapori di riferimento, è facile vedere che i sapori non sono in relazione biunivoca: l'amaro (es. il caffé) è equilibrato dal dolce (zucchero), ma a sua volta il dolce è solo attenuato dall'amaro. Così, se si desidera equilibrare un sapore troppo dolce si aggiunge sale (l'aggiunta è in quantità inferiore rispetto al gusto di partenza, aumentandola fino al sapore desiderato); diversamente, se si addizionasse un amaro avremmo solo un'attenuazione a meno di usare una quantità molto maggiore di amaro rispetto al salato).
Come mostra la tabella, una possibilità da non trascurare è la sostituzione del sale con le spezie. Infatti, il salato viene esaltato dall'amaro e dall'aspro, per cui si può ridurre il sale (per le diete iposodiche), rinforzando la poca quantità usata con sostanze aspre o amare (il pepe - che costituisce una combinazione di aspro e amaro) sulle patate arrosto, oppure limone e/o aceto sull'insalata).
compresse
le compresse sono forme farmaceutiche unidose preparate comprimendo, con apposite macchine, volumi uguali di sostanze solide allo stato di polveri o granuli. Questa forma farmaceutica è notevolmente diffusa, sebbene presenti possibili inconvenienti di tollerabilità ed una biodisponibilità legata al tempo necessario perché si disaggreghi.
Le compresse possono convenientemente essere classificate in base alla loro destinazione:
- compresse semplici o rivestite (confetti): da deglutire come tali;
- tavolette: non devono essere deglutite come tali, ma sciolte in bocca;
- compresse masticabili: devono essere masticate prima della deglutizione;
- compresse sublinguali: vengono assorbite dalla mucosa linguale (p. e. nitroglicerina);
- compresse solubili: devono essere sciolte in acqua, spesso sono effervescenti per favorire la dissolizione o migliorare il sapore;
- compresse sterili per preparare soluzioni iniettabili;
- compresse sterili per impianti sottocutanei;
- compresse da usare come ovuli vaginali, che hanno quasi sostituito gli ovuli di gelatina;
- compresse per praparare soluzioni per uso esterno o diagnostico (test di gravidanza, sterilizzatori per dentiere, ecc.);
- coni dentali
eccipienti per compresse
per trasformare le polveri o i granuli in compresse, occorre addizionare adatti eccipienti che ne favoriscano la compressione, l'ottenimento di un volume adeguato, la disaggregabilità, la compattezza, ecc. In alcuni casi, si può comprimere direttamente la polvere senza che sia stato necessario trasformarla in granulato; tuttavia, questo procedimento è scarsamente applicato in quanto si possono presentare fenomeni di segregazione durante la lavorazione.
Gli eccipienti utilizzati per la preparazione delle compresse, sono classificabili come segue:
- diluenti: aumentano la massa della compressa permettendo il raggiungimento di un volume adeguato per poterla lavorare. Occorre tener presente che, in alcuni casi, i diluenti possono interagire con il farmaco, diminuendone la disponibilità; ad esempio, le tetracicline con l'idrossido di Al, con l'ossido di Mg o con il posfato di Ca;
- adsorbenti: sono utilizzati per aggregare i granuli. Inoltre, si impiegano solventi che, evaporando, esercitano un'azione adesiva (acqua o alcol) ossia soluzioni sciroppose o sostanze collose (gomma arabica, pectine, PVP, metilcellulosa, carbossimetilcellulosa, gelatina, ecc.);
- lubrificanti: migliorano la lavorabilità delle polveri, evitando che si attacchino ai macchinari e riducendo la frizione tra le particelle e lo stampo; infatti, durante la compressione meccanica, l'attrito può essere così elevato da far raggiungere alle polveri temperature prossime a 70 ºC. Si impiegano stearati di Ca, Mg, Al, alcol stearilico, alcol stearico, alcol cetilico, acido palmitico, alcol, amido, PEG, talco.
- glidanti: questi agenti si interpongono fra le particelle della polvere o granulato al quale sono addizionate in modo da riempire le cavità irregolari presenti sulla loro superficie; in questo modo, la forma delle polveri o granulati diviene più regolare e si riduce l'attrito. E' ovvio che i glidanti devono essere finemente suddivisi ed essere aggiunti prima della compressione. La loro efficacia è quantificabile mediante la determinazione dell'angolo di scorrimento o di riposo. Le sostanze utilizzate sono generalmente quelle citate a proposito dei lubrificanti.
- antiadesivi: riducono l'adesione tra compresse e stampo (o punzone), permettendone una facile estrazione. Si impiegano talco, olio di vaselina, acido stearico, stearati di Ca, Mg, ecc.
- disaggreganti: diminuiscono il tempo di disaggregazione delle compresse. Si impiegano derivati dell'amido, alginati, silice, miscele effervescenti, sali molto solubili. Per esempio, l'amido a contatto con l'acqua si rigonfia e disaggrega le compressa; la silice aumenta la capillarità permettendo all'acqua di penetrare più facilmente nella compressa.
Molte sostanze che rientrano nella classificazione esaminata, presentano azione antagonista: per esempio, aggreganti e disaggreganti. Ciò comporta la necessità di raggiungere il miglior equilibrio tra necessità di fabbricazione, biodisponibilità e praticità d'uso. Inoltre, alcune sostanze, pur in diversa misura presentano proprietà glidanti, lubrificanti e antiaderenti: queste sostanze sono dette antifrizione (v. tab. sotto)
|
|
||||
| composto | % di impiego | glidante | antiaderente | lubrificante |
| stearati metallici | 0,5 - 2 | scarsa | buona | ottima |
| talco | 1 - 5 | buona | ottima | scarsa |
| acido stearico | 1 - 4 | nessuna | scarsa | buona |
| cere altofondenti | 3 - 5 | nessuna | scarsa | ottima |
| amido | 5 - 10 | ottima | ottima | scarsa |
produzione delle compresse
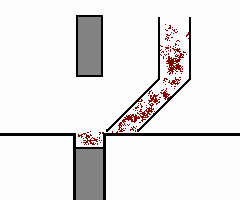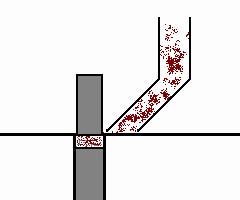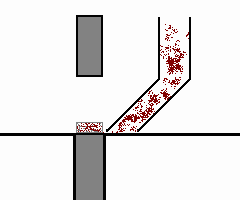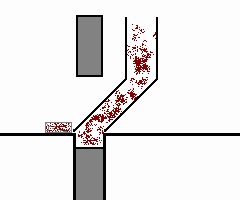 Nell'animazione a fianco è rappresentato schematicamente
il funzionamento di una macchina comprimitrice a punzone singolo.
Nell'animazione a fianco è rappresentato schematicamente
il funzionamento di una macchina comprimitrice a punzone singolo.
In realtà, vengono utilizzate comprimitrici con punzoni multipli, in grado di produrre 250-600 mila
compresse/h. Ovviamente, per ottenere questi risultati, la velocità di funzionamento della macchina deve essere molto elevata, sicché le caratteristiche di scorrevolezza, granulazione, densità,
ecc., delle polveri assumono particolare importanza. In alcuni casi, per raggiungere elevate velocità di lavorazione, gli stampi vengono riempiti sotto pressione.
Con un procedimento simile a quello descritto, si possono preparare anche compresse multistrato, con due o tre strati sovrapposti, usate principalmente nei casi di incompatibilità dei principi attivi. Oppure, compresse rivestite da un nucleo centrale rivestito da una seconda sostanza, per ottenere una compressa a rilascio controllato o gastroresistente. Le compresse rivestite sono prodotte con un processo di "doppia compressione"; questa lavorazione, essendo a secco, è molto meno laboriosa della confettatura ma non sempre si può applicare in quanto il granulato deve possedere particolari caratteristiche di scorrevolezza per permettere un perfetto collocamento sulla prima compressa.
La forma, la dimensione e il diametro delle compresse dipendono, oltre che dalla quantità di sostanza, anche dall'uso al quale la compressa è destinata. Le normali compresse si rompono comprimendole con una forza di 40-80 N, le tavolette resistono a 100-200 N in quanto devono essere succhiate. Le compresse sublinguali sono usate per sostanze che vengono assorbite lentamente e completamente attraverso la mucosa orale (ad es. nitroglicerina, metiltestosterone), in questo caso, gli eccipienti devono essere il più possibili insapori per evitare lo stimolo della deglutizione. Le compresse effervescenti hanno resistenza variabile e, quale requisito principale, è richiesto che non intorbidiscano l'acqua usata per scioglierle; così si utilizzano sostanze tipo NaCl e benzoato sodico, evitando quei lubrificanti, glidanti ed antiadesivi che, essendo insolubili, verrebbero a galla. Le compresse masticabili richiedono eccipienti tipo il mannitolo, che conferisce un'adeguata resistenza unita ad una sensazione di freschezza (dovuta al calore di dissoluzione negativo). Le compresse per impianto, generalmente di forma allungata, hanno elevata resistenza (>200N) e sono utilizzate principalmente per impiantare sottocute sostanze ormonali, con una durata d'azione che può raggiungere un anno.
confettatura
Le compresse possono essere opportunamente rivestite allo scopo di mascherare eventuali odori e sapori sgradevoli, per proteggerle dall'umidità o dall'ossidazione, per controllare il rilascio della sostanza attiva, oppure per ottenere un rivestimento gastroreistente. La confettatura, a volte, può avere il solo scopo di migliorare l'aspetto estetico.
 bassine da 100 kg mod. B100 (ROLLERMAC s.a.s. - www.rollermac.it) |
Il rivestimento può essere realizzato con tecniche diverse. L'attrezzatura base per la confettatura è costituita dalla cosiddetta bassina: un contenitore (in rame o acciao inox) bombato, con
un'apertura per l'introduzione del prodotto da rivestire.
I modelli più grandi, hanno una capacità fino a 250 kg (si deve tener conto che con la confettatura le compresse raddoppano quasi il loro volume). La bassina ruota attorno ad un asse inclinato a
circa 45o; inoltre, sopra l'imboccatura è presente un dispositivo che permette di soffiare aria calda nel recipiente, unitamente ad un sistema aspirante per eliminare la polvere che
può eventualmente formarsi nella fase iniziale della lavorazione.
Prima di esaminare le varie fasi della confettatura, occorre precisare che le compresse da trattare devono avere una forma rotondeggiante senza spigoli vivi; inoltre, devono avere un basso grado di friabilità e devono contenere un disaggregante che favorisca il successivo rilascio nell'organismo.
Fasi della lavorazione:
- gommatura: serve per evitare il contatto diretto tra le compresse e le successive soluzioni acquose (v. punto 2). Si effettua sciogliendo in un solvente organico (comunemente alcol) sostanze quali gomma lacca, aceto-ftalato di cellulosa, zeina (proteina), oppure una loro miscela; questa soluzione viene introdotta nelle bassina e fatta miscelare con le compresse. L'alcol viene eliminato mediante immissione di aria calda e contemporannea aspirazione; inoltre, per evitare che le compresse possano attaccarsi tra loro, si aggiunge un pò di talco, il cui eccesso viene poi aspirato. Durante la gommatura, di solito occorre effettuare due passaggi.
-
prerivestimento: viene effettuato in vari stadi. Consiste nell'aggiungere alternativamente sostanze sciroppose (zucchero 150 g, gomma arabica, 6 g, acqua depurata 100 g)
e polveri (calcio carbonato 35 g, caolino 15 g, talco 25 g, zucchero 20 g, gomma arabica 4g).
Si procede in questo modo: dopo il trattamento con la soluzione sciropposa, si fa' solidificare lo strato zuccherino che si è depositato sulle compresse, in modo che resti ancóra molliccio e adesivo in modo che la successiva polvere aderisca più facilmente; questo trattamento alternato, viene ripetuto più volte finché la compressa assume una forma tondeggiante. - ingrossamento: si effettua solo con la soluzione zuccherina (senza alternare con polveri) in cui eventualmente si può inglobare un pò di calcio carbonato ed amido; anche questo processo va ripetuto varie volte impiegando ogni volta una soluzione un pò più diluita per rendere il confetto più regolare.
- finitura e lisciaggio: si esegue usando solo sciroppo semplice diluito (senza altri componenti). In questa fase finale si possono eventualmente aggiungere allo sciroppo sostanze opacizzanti e coloranti (analoghe a quelle usate per le capsule).
- lucidatura: si effettua in un altro tipo di bassina non bombata ma a forma di tamburo e con le pareti interne rivestite in tela. In questo recipiente si aggiunge una soluzione di cera d'api o cera carnauba in un solvente volatile organico volatile. I confetti, rotolando in questa soluzione, si ricoprono con un sottile strato di cera che li rende lucidi e poi, dopo l'evaporazione del solvente, la leggera azione abrasiva della tela completa l'opera di lucidatura della cera.
rivestimenti gastroresistenti e per il rilascio controllato
Si ricorre a questo rivestimento se il principio attivo è alterabile dai fluidi gastrici, oppure perché ragioni farmacologiche richiedono che debba essere assorbito solo a livello intestinale, oppure per mantenere più a lungo la durata dell'azione.
La tipologia delle sostanze usate in questi rivestimenti varia a seconda che si voglia ottenere solo un'azione ritardata o anche gastroresistenza; per quest'ultima, occorre utilizzare sostanze
non degradabili nell'ambiente acido dello stomaco, ma solo in quello neutro o alcalino dell'intestino. Le tecniche di lavorazione prevedono sempre l'uso di un solvente organico che evaporando
lascia una pellicola dotata delle necessarie proprietà.
Le pellicole di rivestimento possono essere di due tipi:
- con la sola funzione di schermo meccanico per rallentare il rilascio del principio attivo. A questo scopo, si usano derivati della cellulosa (ossi-propil-etil-cellulosa, ecc.) oppure i PEG e
derivati, ed il PVP.
- gastroresistenti, che si sciolgono solo nell'intestino. Si usano sostanze aggredibili o dagli enzimi o dall'ambiente intestinale. Generalmente, queste sostanze contengono gruppi esteri che vengono idrolizzati nell'intestino, ma non nello stomaco; per es., si usa il fenile salicilato (salolo) che è un estere idrolizzabile solo a pH alcalino o neutro, oppure alcune cere sempre di natura esterea. Queste sostanze possono essere idrolizzate soprattutto per azione di particolari enzimi (lipasi) capaci di scindere il legame estere. Sono utilizzate anche sostanze di tipo proteico (ad es., la cheratina), che subiscono l'attacco di enzimi proteolitici attivi a pH > 6. Queste sostanze presentano scarsa solubilità in acqua e per potere rivestire le compresse vengono sempre usate soluzioni con solventi organici facilmente eliminabili. Un'apparecchiatura abbastanza utilizzata per questa operazione è simile a quella utilizzata nella polverizzazione per nebulizzazione: le compresse, inizialmente alloggiate nella griglia che costituisce il fondo del recipiente, vengono sollevate da un getto d'aria calda e quindi, mentre circolano sospese in un vortice, sono investite da uno spruzzo costituito dalla soluzione di rivestimento. Regolando opportunamente il flusso del getto d'aria, che ha anche la funzione di far evaporare il solvente, si può fare in modo che quando le compresse sono sufficientemente ricoperte, il loro peso diventi tale da farle cadere sul fondo. A questo punto, la lavorazione è terminata. Oltre a questa tecnica, che richiede una notevole quantità di energia ed è comunque adatta solo per compresse particolaremnte resistenti, si può usare quella descritta per la bassina.
capsule
le capsule sono forme farmaceutiche monodose per uso orale o suppositorio, all'interno delle quali si possono introdurre granulati, polveri o miscele oleose.
Le capsule sono costituite essenzialmente da gelatina, da un agente plasticizzante (glicerina, sorbitolo, sciroppo semplice, amido), da sostanze conservanti (ad es. piccole quantità di anidride solforosa per prevenire la crescita di muffe e batteri sulla gelatina che costituisce un ottimo terreno di coltura), e da coloranti naturali e opacizzanti (in assenza di questi ultimi, generalmente biossido di titanio, la capsula è trasparente). Nel caso si vogliano capsule gastroresistenti, si ricorre alla cellulosa acetoftalato (un polimero enterosolubile).
I farmaci sono a volte confezionati sotto forma di capsule o
di compresse laccate per coprire un sapore particolarmente sgradevole, per contenere una sostanza fluida o per proteggere il principio attivo dall'azione dei succhi gastrici: è pertanto
importante non frantumare le compresse e non aprire le capsule, a meno che sia esplicitamente segnalato sul foglietto illustrativo.
Per esempio, gli inibitori dell'ATPsintasasi (o ATPasi) e gli inibitori della secrezione gastrica di acido cloridrico (omeprazolo, lansoprazolo, pantoprazolo e rabeprazolo) sono formulati in
capsule che rilasciano il principio attivo solo nell'intestino, dove viene assorbito, in quanto se venisse a contatto con i succhi gastrici sarebbe completamente inattivato. Quindi è obbligatorio
ingerire le capsule intere, senza aprirle o romperle, pena l'inefficacia del farmaco. Anche per i fermenti lattici, vale la stessa avvertenza: quando non sono protetti da capsule
gastroresistenti, il numero dei batteri che arrivano all'intestino è ridicolmente basso.
produzione delle capsule
 La differenza principale tra le capsule molli e le capsule dure (dette anche
opercoli), consiste nel fatto che le prime vengono oggi preparate con un unico procedimento e non è possibile aprirle se non tagliandole o perforandone l'involucro; le seconde, invece, vengono
preparate vuote e vendute come tali all'industria utilizzatrice che le riempie con apposite macchine (v. animazione a destra) che permettono un riempimento fino a 150.000 cps/h, e possono
stampigliare sulla capsula il nome della specialità e dell'officina.
La differenza principale tra le capsule molli e le capsule dure (dette anche
opercoli), consiste nel fatto che le prime vengono oggi preparate con un unico procedimento e non è possibile aprirle se non tagliandole o perforandone l'involucro; le seconde, invece, vengono
preparate vuote e vendute come tali all'industria utilizzatrice che le riempie con apposite macchine (v. animazione a destra) che permettono un riempimento fino a 150.000 cps/h, e possono
stampigliare sulla capsula il nome della specialità e dell'officina.
Il fatto che le industrie utilizzino capsule dure già pronte è conseguente al fatto che la loro preparazione
richiede una tecnologia complessa non facilmente ammortizzabile in termini di costi. Infatti, sono solo tre ditte (e loro consociate) che producono capsule per tutte le officine
farmaceutiche.
La preparazione delle miscele da allestire in capsule di gelatina molle è un'operazione completamente diversa. L'unica fase della preparazione dei lotti durante la quale le sostanze sono alla
stato di polvere, è la pesatura. Poiché le capsule di gelatina molle vengono quasi sempre riempite con liquidi, oppure con sostanze a consistenza pastosa, le polveri appena pesate vengono
addizionate con opportuni eccipienti liquidi, che sono normalmente olii vegetali oppure minerali, oppure ancóra eccipienti liquidi come il polietilenglicole. Ne risulta una pasta che viene
omogeneizzata con apparecchi di vario tipo. La miscela viene poi setacciata e messa sotto vuoto in modo che la pasta risultante non contenga bolle d'aria che potrebbero procurare inconvenienti
nell'esattezza del dosaggio.
L'omogenizzazione di una miscela in fase fluida (sospensioni o emulsioni) è ovviamente molto più facile a realizzarsi rispetto ad una miscela in fase solida. Per evitare la sedimentazione dei
componenti più pesanti, sarà sufficiente addensare o aumentare la viscosità dell'eccipiente liquido, e per assicurare una perfetta distribuzione di tutti i componenti,sarà sufficiente influire
leggermente sulla tensione superficiale del liquido in modo che tutti i granelli vengano bagnati omogeneamente e non si formino grumi.
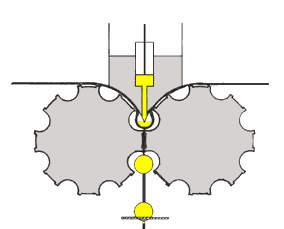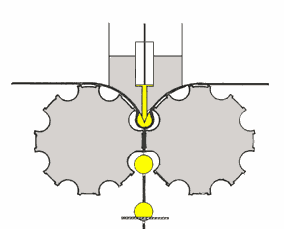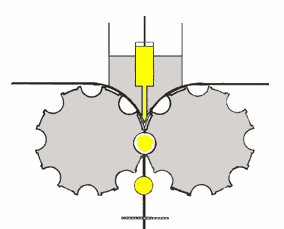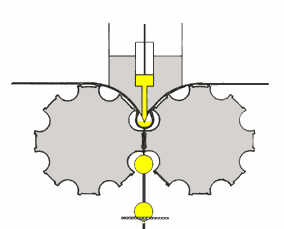 Lo schema del processo d'incapsulazione
(brevetto Scherer s.p.a.), che in questo caso necessita di un commento, è riportato nell'animazione a fianco.
Lo schema del processo d'incapsulazione
(brevetto Scherer s.p.a.), che in questo caso necessita di un commento, è riportato nell'animazione a fianco.
Da due serbatoi di gelatina (non in figura), mantenuti ad una temperatura opportuna, viene deposta una pellicola su due tamburi rotanti (non in figura) posti simmetricamente. Le due pellicole,
mantenute in tensione da due cilindri lubrificati (non indicati in figura), scorrono su due matrici cilindriche contrapposte sulle quali sono incise scanalature (alveoli) a forma di mezza capsula
(la forma e il volume degli alveoli ppossono essere variate sostituendo le matrici). A questo punto, gli ugelli collegati ad una pompa volumetrica iniettano simultaneamente le dosi di farmaco
(corrispondenti al volume di ogni alveolo) in ciascuna capsula nel momento che le due pellicole di gelatina si toccano. Il preparato spinge così la gelatina dentro gli alveoli; la capsula viene
quindi saldata e poi tagliata (non in figura). Le capsule sono infine sgrassate ed essiccate in corrente d'aria calda.
 particolare della banda sigillante tra le due parti della capsula (foto tratta da Shionogi qualicaps www.qualicaps.com) |
 particolare dell'applicazione della banda sigillante (foto tratta da Shionogi qualicaps www.qualicaps.com) |
Anche le capsule dure permettono di contenere fluidi; per questo, un'apposita macchina applica una banda di gelatina a bassa viscosità che è depositata tra le due parti (superiore ed inferiore) della capsula. Se si presenta qualche bolla, questa è rapidamente eliminata da una seconda applicazione della soluzione sigillante. La seconda applicazione è più sottile della prima, producendo un legame uniforme e stabile. Lo strato è poi essiccato ad aria, evitando qualsiasi esposizione termica che potrebbe produrre bolle e causare fragilità nel rivestimento di gelatina. La fragilità può comportare perdite durante le operazioni di trasporto alle linee di confezionamento, senza dimenticare la frustrazione del fruitore quando nel rimuovere le capsule dal blister queste si rompono o si aprono.
La banda sigillante garantisce alcuni benefici:
- prevenzione della perdita del riempimento;
- prevenzione della permeabilità all'ossigeno;
- garanzia che le capsule non si aprano durante la deglutizione, prevenendo infiammazioni all'esofago;
- prevenire la manomissione della banda di chiusura.
Il riempimento di capsule dure con formulazioni liquide non è senza problemi. L'uso di iniettori volumetrici costituisce certamente uno dei più accurati mezzi di dosaggio disponibili; d'altra parte, la lavorazione di una capsula riempita con un liquido a bassa viscosità richiede accorgimenti particolari che non si presentano con formulazioni in polvere. Un movimento intermittente può produrre spargimento quando sono raggiunte elevate velocità. Inoltre, l'azione di pompaggio idraulico che si crea quando la parte superiore è applicata alla capsula riempita di fluido può produrre una chiusura non perfetta.
capsule ad effetto protratto
E' possibile preparare medicinali ad azione protratta formulati come capsule di gelatina. Nel caso di capsule
di gelatina dura, queste possono essere riempite con cronoidi (diffucaps) o microcapsule a cessione protratta.
Nel caso delle capsule di gelatina molle, oltre alla possibilità di riempirle con cronoidi e microcapsule,
eventualmente sospesi in eccipienti liquidi, è possibile aggiungere agli eccipienti delle sostanze che, a contatto con i succhi gastrici o enterici, rallentino la disgregabilità della capsula
ritardando la cessione dei principi attivi.
riempimento delle capsule per preparazioni estemporanee
|
|
|
| misura della capsula | volume approssimato |
| 00 | 0.95 ml |
| 0 | 0.70 ml |
| 1 | 0.50 ml |
| 2 | 0.40 ml |
| 3 | 0.30 ml |
| 4 | 0.20 ml |
| 5 | 0.15 ml |
 opercolatrice da laboratorio: il riempimento avviene disponendo la parte inferiore degli opercoli nelle apposite sedi; quindi si distribuisce la polvere rasa (ecco la necessità del calcolo delle dimensioni delle capsule) e si procede alla chiusure. |
Le capsule opercolate vuote sono commercializzate con capacità diverse. Le capsule più comunemente usate variano dal numero "00" al numero "5" (il più piccolo). La capacità delle capsule (nella tab. accanto) è indicativa in quanto il loro volume è riferito ad un liquido non acquoso, per esempio alcol. Il peso di polvere che può essere contenuto in una capsula dipende dal volume apparente della polvere (varia con il grado di umidità, la scorrevolezza e la granulometria) e può variare a seconda della pressione esercitata per riempire la capsula.
Per fissare le idee, supponiamo di voler preparare trenta capsule contenenti 300 mg di acido acetilsalicilico e 50 mg di caffeina. Per far questa preparazione, si miscelano le due polveri con il
metodo della diluizione progressiva (v. nota in basso), si trasferiscono in un cilindro graduato e si legge il volume occupato dalla miscela (volume apparente), per esempio 19 ml : per ogni dose,
avremo quindi 19/30 = 0.63 ml. Questo significa che dovremo utilizzare capsule misura "0" con capacità 0.70 ml. Ora, per preparare materialmente le capsule, occorre raggiungere il volume di 0.70 ml
per capsula = 0.70 x 30 = 21 ml : aggiungiamo eccipiente in leggero eccesso fino ad ottenere 21.5 ml; quindi travasiamo la miscela nel mortaio e completiamo la
miscelazione.
diluizione progressiva: questo procedimento
garantisce una efficace distribuzione omogenea fra due polveri. Supponendo di voler miscelare 50 g di polvere A con 10 g di polvere B, si opera in queto modo: si
prende la polvere (in quantità minore) B e si addiziona con una eguale quantità di A (10 g di B e 10 g di A); poi si
aggiunge alla miscela una seconda quantità uguale di A (20 g di A + B e 20 g di A); infine si aggiunge il
residuo A alla miscela.
preparazioni iniettive
Le soluzioni iniettabili possono essere costituite da soluuzioni, sospensioni o emulsioni sterili, confezionate in contenitori chiusi e destinate ad essere somministrate per via sottocutanea, intramuscolare o endovenosa.
I principali controlli ai quali devono soddisfare le preparazioni iniettive (per i saggi specifici si rimanda alla F.U.), ne definiscono anche le carattteristiche.
- assenza di particelle estranee: le cause più frequenti di inquinamento possono derivare da cessione del vetro, del tappo (per quelli perforabili), dai processi di saldatura delle fiale, da modificazioni chimico-fisiche del preparato;
- pressione osmotica: deve essere compatibile con i liquidi tissutali con cui entrerà in contatto; questa condizione è particolarmente importante per le preparazioni endovenose;
- concentrazione idrogenionica: il pH riveste un ruolo importante in quanto è legato alla tollerabilità dell'organismo ed alla stabilità del farmaco. In generale, preparazioni con pH compreso fra 4 e 10 sono discretamente tollerate dal sangue e dai tessuti in conseguenza dei sistemi tamponanti naturali. Per le sostaze chimicamente instabili, si può, tuttavia, ricorrere a miscele tampone (ad es. acido citrico/citrato trisodico - pH = 3.6; ac. acetico/acetato di sodio - pH = 3.6-5.6; carbonato monosodico e disodico - pH = 9.2-10.7);
- assenza di sostanze pirogene e sterilità: queste condizioni sosno strettamente legate in quanto le sostanze pirogene sono tossine di origine generalmente batterica.
 I veicoli usati per le preparazioni iniettabili sono l'acqua e gli olii
vegetali (di oliva, di sesamo, di mandorle dolci, di arachidi o di semi di cotone) e devono mantenere le loro caratteristiche chimico-fisiche anche dopo la sterilizzazione.
I veicoli usati per le preparazioni iniettabili sono l'acqua e gli olii
vegetali (di oliva, di sesamo, di mandorle dolci, di arachidi o di semi di cotone) e devono mantenere le loro caratteristiche chimico-fisiche anche dopo la sterilizzazione.
Si usano anche veicoli quali la glicerina, il polietilenglicole, il glicole polietilenico, l'oleato di etile.
Per aumentare la stabilità ed assicurare la sterilità, si possono aggiungere additivi (tamponi, agenti solubilizzanti, antiossidanti, antibatterici): devono essere attivi a bassa concentrazione,
non devono interferire con il farmaco né influenzare i suoi saggi specifici.
preparazioni per via endovenosa
I farmaci destinati alla somministrazione endovena devono essere idrosolubili alla concentrazione attiva: se il farmaco è solubile in acqua si usano soluzioni acquose isotoniche (se è instabile in acqua si usa una polvere da solubilizzare al momento dell'uso): se è insolubile, si può cercare di ottenere modificazioni molecolari che lo rendano solubile senza alterarne le proprietà farmacologiche.
Si definisce isotonica una pressione osmotica pari a quella esercitata dal liquido extracellulare attraverso le membrane cellulari. La concentrazione isotonica è solitamente riferita al sangue umano: l'isotonia rispetto ai globuli rossi è prodotta da una soluzione allo 0.9% di NaCl in acqua (soluzione fisiologica). Poiché le membrane biologiche non sono ideali, una concentrazione iso-osmotica col sangue non produrrà necessariamente una pressione osmotica isotonica e viceversa.
La tabella seguente (F.U.) elenca le sostanze con le loro rispettive concentrazioni (osmol/L) isosmotiche ed isotoniche rispetto ai globuli rossi umani ed alla congiuntiva umana. Queste soluzioni possono essere usate come veicolo isotonico anche per i medicamenti di cui non si conoscano le caratteristiche di diffusibilità attraverso le membrane cellulari e quindi la pressione osmotica esercitata rispetto alle cellule con cui le soluzioni (da esse prodotte) verranno in contatto. Infatti, se un farmaco non è diffusibile attraverso la membrana cellulare, la soluzione risultante è ipertonica e quindi generalmente ancóra tollerata dal punto di vista osmotico (fino a 2 - 3 volte l'isotonica). Se invece il farmaco diffonde attraverso la membrana cellulare, si ottiene evidentemente una soluzione isotonica.
| sostanza | P.M. | iso-osmot. | isotonica |
| sodio cloruro | 58.44 | 0.90 | 0.91 - 0.93 |
| sodio bicarbonato | 84.01 | 1.39 | 1.3 - 1.4 |
| sodio fosfato bisodico | 358.14 | 4.62 | 3.2 - 3.4 |
| sodio citrato | 294.10 | 3.44 | 2.6 -2.8 |
| sodio solfito | 322.19 | 3.65 | 3.4 - 3.8 |
| calcio cloruro | 147.07 | 2.50 | 2.4 - 2.6 |
| calcio lattato | 308.30 | 6.36 | 6.2 - 6.5 |
| glucosio | 198.17 | 5.51 | 5.5 - 6.5 |
| saccarosio | 342.30 | 9.25 | 9.0 - 9.5 |
| mannite | 182.17 | 5.07 | 5.0 - 5.1. |
In generale, è necessario ricordare che i danni alle cellule animali non sono dovuti
esclusivamente agli insulti osmotici. Perciò, quando si mette a punto una nuova formulazione, è sempre opportuno e, in molti casi indispensabile, verificare sperimentalmente la tollerabilità
della nuova preparazione rispetto al sangue, alla congiuntiva o agli altri tessuti con cui la suddetta preparazione è destinata a venire in contatto.
nanosospensioni per iniezione endovenosa
La somministrazione iv permette un accesso diretto al torrente circolatorio, ottenendo così un rapido inizio dell'effetto terapeutico (e di eventuali rezaioni avverse). Però, le formulazioni iv, oltre ai requisiti discussi in precedenza, non devono contenere particelle solide aventi dimensioni superiori a 5 mm, in modo da garantire un certo margine per evitare l'occlusione dei capillari polmonari (6mm) durante il loro attraversamento. Questa è la ragione per cui nella pratica corrente le preparazioni iv sono essenzialmente soluzioni.
|
| la fig a destra rappresenta schematicamente i fenomeni di sintering eripening di Ostwald. a) sintering: particelle individuali si combinano in un aggregato delimitato da interfacce che le separano le une con le altre; b) ripening: varie particelle si agglomerano in un insieme unico. Entrambi i processi avvengono con riduzione della superficie delle particelle disperse. |
Nel caso di farmaci poco solubili in veicoli acquosi (oppure nei casi in cui la salificazionenon sia consigliata), si può ricorrere a cosolventi (alcol e glicoli) o tensioattivi che possono permettere di solubilizzare il farmaco alla necessaria concentrazione. D'altra parte, questa alternativa presenta problemi legati alla tossicità dei cosolventi e segnatamente dei tensioattivi. Proprio i tensioattivi sono responsabili di fenomeni irritativi e infiammatori localizzati nel punto d'iniezione o a carico dei vasi venosi (flebiti). Per questa ragione, particolarmente per i farmaci destinati ad una terapia prolungata nel tempo, tali formulazioni sono considerate di nicchia.
Un'alternativa all'uso di cosolventi e tensioattivi è costituita dai sistemi colloidali costituiti da emulsioni o liposomi veicolanti il farmaco insolubile in acqua. Tra i sistemi colloidali si possono includere le nanosospensioni: dispersioni in adatto veicolo di particelle solide con dimensioni medie non superiori a qualche centinaio di nanometri (ben sotto la soglia dei 6mm). Particolarmente le formulazioni in nanotecnologia, però, hanno uno svantaggio rispetto alle emulsioni o le micelle lipidiche: la loro struttura non rigida, infatti, ne consente una certa deformazione, permettendo loro di attraversare anche capillari di sezione inferiore al proprio diametro. Un altro problema delle nanosospensioni è la reciproca aggregazione delle particelle, fenomeno che si può contrastare con agenti tensioattivi per formulazioni iniettive (es. fosfolipidi, poloxomeri e polisorbato 80). Questa aggregazione nota come "Ostwald sintering e ripening" è illustrata schematicamente nella figura a fianco.
I principali aspetti critici connessi con la somministrazione iv di nanosospensioni sono costituiti da embolia polmonare, irritazione vascolare, flebiti, ecc. Ed a questi problemi si aggiunge la possibile formazione di polimorfi durante il processo produttivo; inoltre la sterilizzazione mediante filtraggio non può essere applicata per la possibile occlusione dei filtri.
preparazioni parenterali ad azione ritardo
Per modificare la durata dell'azione terapeutica, si può ricorrere a tecniche di somnministrazione, mezzi farmacologici, chimici e tecnologici.
- tecniche di somministrazione: la via intramuscolare e la via sottocutanea garantiscono un certo ritardo rispetto alla via endovenosa;
- mezzi farmacologici: per esempio, si può ricorerre a vasocostrittori, che limitando l'afflusso di sangue nel sito dell'iniezione, prolungano il tempo di rilascio (è un metodo usato soprattutto negli anestetici locali). Altre possibilità, di carattere generale, sono state precedentemente discusse (v. farmacocinetica);
- mezzi chimici: quando possibile, si può ricorrere all'esterificazione o salificazione in sali poco solubili, oppure alla formazione di complessi;
- mezzi tecnologici: sono quelli che più ci interessano. L'assorbimento è ritardato da veicoli oleosi, che possono anche essere strutturati, ad es. con sterarato di alluminio, in modo da trasformare il veicolo in gel tissotropico (per agitazione, il gel diventa sol e può essere iniettato; poi, nel muscolo ridiventa gel). Altri esempi sono offerti dalla varie forme ritardo in complessi dell'insulina. Infine, si possono usare sostanze adsorbenti quali l'idrossido di alluminio, soprattutto per alcuni vaccini.
Per concretizzare le idee, citiamo due specialità medicinali che permettono considerazioni di carattere generale:
CELESTONE CRONODOSE®: è costituito da betametasone fosfato disodico e betametasone acetato in veicolo acquoso. Il betametasone fosfato disodico presente come fase soluta, garantisce una pronta risposta terapeutica. Il betametasone acetato presente in sospensione come fase dispersa, permette un effetto protratto per qualche giorno.
DEPO MEDROL®: è costituito da metilprednisolone acetato in sospensione acquosa. In questo caso, i livelli terapeutici si mantengono per circa 10 gg e quindi, l'impiego terapeutico è adatto al trattamento di fatti infiammatori non episodici (rinite allergica, vasomotoria, ecc.) o cronici.
Entrambe le specialità sono sostanze ormonali: in questo caso, il rilascio del farmaco è parzialmente modulato dal ritmo circadianodi secrezione surrenalica, che prolunga ulteriormente la permanenza nell'organismo. D'altra parte, in caso di problemi di carattere clinico, le forme ritardo hanno lo svantaggio di non permettere l'immediata sospensione dl trattamento (fanno eccezione i sistemi transdermici); in questo caso, si può, per quanto possibile , con limitati risultati, ricorrere a mezzi che aumentino l'escrezione renale per accelerare l'eliminazione del farmaco.
colliri
Si intende per collirio (dal greco kollurion = unguento) qualsiasi preperazione medicamentosa ad uso topico congiuntivale, ossia destinata ad essere applicata sulla mucosa congiuntivale dell'occhio. I colliri sono generalmente liquidi (soluzioni acquose, oleose o sospensioni), ma possono anche essere solidi (pomate, unguenti, polveri).
Dal punto di vista farmacologico, la penetrazione dei princìpi attivi contenuti nei colliri, avviene essenzialmente attraverso la ricca vascolarizzazione del sacco congiuntivale.
La penetrazione attraverso la cornea è invece più selettiva ed è influenzata da vari fattori sui quali non si soffermeremo.
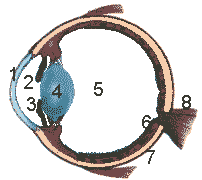 1. cornea: prima lente dell'occhio, molto trasparente, posta davanti all'iride; 1. cornea: prima lente dell'occhio, molto trasparente, posta davanti all'iride;2. umor acqueo: il liquido che conferiece pressione all'occhio; 3. iride: il diaframma colorato dell'occhio, delimita la pupilla; 4. cristallino: seconda lente dell'occhio (focalizzazione); 5. umor vitreo: un "gel" di sostegno; 6. retina: è la parte nervosa, il supporto che raccoglie i segnali ottici; 7. sclera: la struttura di sostegno; 8. nervo ottico: il collegamento al cervello; 9. congiuntiva: ricopre la sclera (non disegnata in figura) |
Come eccipienti per colliri, si possono impiegare la vaselina e l'olio d'oliva, addizionati con lanolina per incorporarvi un pò d'acqua; si possono anche utilizzare polietilenglicoli o emulsioni A/O o O/A. Per gli emulsionanti, i non ionici presentano una migliore tollerabilità.
Inoltre, come requisiti essenziali, sono richiesti:
- un pH tollerabile (generalmente compreso tra 6.4 e 7.8);
- sterilità ed assenza di particelle estranee;
- pressione osmotica isotonica con le lacrime o ipertonica (cioè con pressione osmotica corrispondente a quella prodotta da soluzioni di NaCl dallo 0,6 al 2,7%). Va ricordato però che le osmolarità molto basse (<150 mosm/l) possono dimostrarsi irritative per l’epitelio corneale.
- per quanto riguarda l'assorbimento, gli antisettici devono svolgere esclusivamente azione superficiale.
liquido lacrimale
Il liquido lacrimale è formato per il 98% circa da acqua e per il 2% da sostanze varie, tra le quali predomina il cloruro di sodio. Inoltre, nel liquido lacrimale è presente il lisozima, un componente la cui azione battericida conferisce al liquido un certo potere disinfettante, a salvaguardia del globo oculare dalle infezioni microbiche. La produzione del liquido lacrimale, da parte delle omonime ghiandole, avviene ininterrottamente, assolvendo così a tre importanti funzioni:
- lava il globo oculare, mantenendolo sempre terso e pulito dal pulviscolo atmosferico e da minuscoli corpuscoli estranei, con conseguente nitidezza della visione oculare;
- umetta in permanenza il globo oculare, che altrimenti sarebbe destinato ad irruvidirsi a contatto con l'aria;
- contrasta l'attecchimento dei microbi patogeni sul globo oculare, riducendone la possiilità di infezioni.
Da quanto riassunto, risulta evidente l'importanza che il collirio, nella cui formulazione sono compresi tensioattivi e conservanti, non interferisca con la lacrimazione.
trattamento dell'occhio secco
L'occhio secco non è un problema legato all'inquinamento ambientale; infatti, già nella Grecia antica si usavano impacchi d'albume d'uovo e grasso d'oca. I Tuareg utilizzavano piccoli semi di Hydrofilia Senegalesis che inseriti nel sacco congiuntivale dissolvevano una mucillagine utile a contrastare il forte e caldo vento del deserto.
Normalmente il pH lacrimale è attorno a 7,2-7,4. Il paziente riferisce sensazione di benessere quando il collirio è alcalino. Normalmente le lacrime artificiali (v. avanti) sono a pH tamponato. È possibile trovare anche colliri a pH variabile; tuttavia, occorre sottolineare due punti:
- i colliri acidi rispettano l'impalcatura della struttura mucoide;
- i colliri alcalini hanno funzione mucolitica e possono quindi essere utili per correggere gli eccessi di muco coagulato.
osmolarità nell’occhio secco: l'osmolarità normale è di 303-305 mosm/l. Nelle CCS (cheratocongiuntiviti secche) la lacrimazione diventa
iper-tonica, fino a raggiungere osmolarità di 340 mosm/l.
Anche in molti altri casi di iper-evaporazione, derivanti dall'uso di LAC (lenti a contatto), esposizione a
VDT (videoterminali, riduzione del lipidico), l'osmolarità può aumentare ma in misura inferiore.
Va ricordato però che le osmolarità molto basse (<150 mosm/l) possono dimostrarsi irritative per
l'epitelio corneale.
Lacrime ipertoniche sono invece consigliate nella cheratite filamentosa oppure nell'edema epiteliale corneale
e nelle frequenti erosioni corneali recidivanti.
tensione superficiale nell'occhio secco: una delle funzioni fondamentali dei
sostituti lacrimali è quella di ripristinare una normale tensione superficiale. Si tratta della capacità propria dello strato mucoso di permettere alla fase acquosa di distendersi
sull'epitelio.
La tensione superficiale dei colliri per soddisfare tale funzione dovrebbe aggirarsi teoricamente attorno a 42 dyne/cm (tensione superficiale del muco idrofilico oculare sull'epitelio corneale).
Quella della fase acquosa sulla fase mucinica è di circa 6 dyne/cm. I migliori sostituti lacrimali oggi garantiscono valori attorno a 50 dyne/cm.
lacrime artificiali
Da quanto esposto, è ovvia l'importanza della corretta lacrimazione dell'occhio: nei casi di secchezza dovuta a limitata lacrimazione, si può ricorrere a soluzioni antisettiche e lubrificanti.
Le lacrime artificiali sono soluzioni saline normalmente al 0,9% di NaCl e KCl tamponata al 7,2-8,0 pH. La funzione di queste soluzioni è fondamentalmente umidificante e mucomimetica
(mimano le funzioni dello strato mucoproteico).
-
le lacrime artificiali devono essere:
- soluzioni ben tollerate;
- soluzioni atossiche, anche se applicate con frequenza;
- soluzioni con periodo di ritenzione lungo in sede locale;
- non devono influenzare la visione;
- non devono ostacolare le normali secrezioni muco-lipidiche-acquose;
- non devono ostacolare l’osmolarità corneale e il suo metabolismo;
- non devono emulsionare i lipidi;
- devono possedere la minor concentrazione possibile di conservanti.
La funzione umidificante è garantita dalla soluzione acquosa isotonica contenente i normali elettroliti lacrimali.
-
i componenti mucomimetici devono:
- garantire l'idrofilìa dell'epitelio;
- assicurare la cattura degli inquinanti lipidici e renderli idrofilici;
- proferire viscosità alla lacrimazione;
- permettere allo strato lipidico di disporsi rapidamente sullo strato acquoso.
Poiché sostanze mucomimetiche di provenienza animale o vegetale si sono rivelate inutilizzabili, si ricorre a
soluzioni contenenti polimeri idrofili.
Tali soluzioni si possono presentare a diversa viscosità. Occorre però rimanere entro certi limiti per
evitare alterazioni della visione. Di conseguenza la loro ritenzione sull'occhio è limitata. Pazienti con disturbi gravi o moderati dell'idratazione devono utilizzare tali preparati con
frequenza.
Due fattori importanti ed ineliminabili, diminuiscono l'efficacia delle lacrime artificiale, con conseguente notevole riduzione della capacità umettante e umidificante della lacrima artificiale:
- lacrimazione riflessa (ulteriore diluizione della soluzione);
- ammiccamento riflesso creato dall'applicazione del collirio sul sacco congiuntivale.
A 90 secondi dall'applicazione nel sacco congiuntivale di pazienti in cui era stato instillato il prodotto, si sono ritrovati solo:
- 3% di soluzione fisiologica,
- 5% di soluzione alcool polivilinico,
- 10% di soluzione metil cellulosa.
Secondo gli studi, il BUT (Break Up Time = tempo di rottura del film lacrimale)- dopo instillazione di lacrime artificiali - può incrementare e rimanere stabile fino a 120 minuti.
Come lacrime artificiali, si possono utilizzare:
metilcellulosa: usata fin dal 1945 può essere utilizzato a varie concentrazioni dallo 0,5 all’1%. E' atossica, inerte, con pH stabile. Ha un indice di rifrazione (a
concentrazione 1%) simile al quello lacrimale.
Per 10-20’ ha buon effetto lubrificante. Incrementa il BUT di un fattore pari a 1,4 (questo valore potrebbe aumentare se si portasse la concentrazione della metilcellulosa a 1,5%, ma si
preferisce evitare per via degli effetti di intorpidimento della visione e delle crosticine sulle ciglia).
A concentrazioni elevate le soluzioni contenenti metilcellulosa rallentano i processi di cicatrizzazione delle lesioni corneali.
Rispetto agli altri elementi umettanti, la metilcellulosa è la sostanza che più rimane in sede. Per questo viene usata nei casi medio gravi di ipo-secrezione o alterazine del
lipidico-mucinico.
Dalle modifiche strutturali della metilcellulosa derivano: idrossimetilcellulosa e carbossimetilcellulosa.
idrossipropilcellulosa: È un derivato della cellulosa con migliori proprietà di superficie. E' considerato relativamente migliore degli altri preparati di cellulosa. Dà buona soluzione ai sintomi di secchezza oculare. Unico neo, la tensione superficiale alta: 60 dyne/cm che aumenta il BUT ma riduce la bagnabilità.
alcool-polivilinico: la concentrazione mediamente usata varia dall'1,4 al 3% (è atossico anche a concentrazioni del 10%). Ha buona funzione viscosante ma soprattutto
idratante. Ha una buona tensione superficiale 46 dyne/cm e quindi costituisce una buona soluzione a fini idratanti. L’alcool polivilinivo possiede alcune proprietà simili a quelle dello strato
mucinico: riduzione della tensione lacrime/aria e della tensione lacrime/epitelio. Ha buona funzionalità in soluzioni a pH alcalino.
Dagli ultimi studi risulta che il tempo di ritenzione a livello oculare è leggermente maggiore rispetto a quello dei derivati della cellulosa. E' un buon sostituto dello strato mucinico e
acquoso.
gel lacrimali
Sono sostanze gelatinose a base poliacrilica. Il loro grande vantaggio è quello di distribuirsi sulla superficie oculare senza formare striature. Gli studi hanno dimostrato la capacità di aumentare il BUT (Break Up Time = tempo di rottura del film lacrimale; viene determinato con un esame oculistico) fino a 60’. Rispetto all'alcool polivilinico rimane in sede per un fattore pari a 7. L’unico problema connesso all’uso dei gel oculari è la tendenza ad accumularsi sui bordi palpebrali, dove si secca e forma crosticine.
agenti viscosanti
acido ialuronico: è una sostanza organica presente nei tessuti di tutti i vertebrati. È dotato di un'elevata proprietà viscoelastica.
È in grado di modificare la propria viscosità a seconda delle forze di frizionamento cui è sottoposto (come le lacrime naturali).
Grazie alla buona capacità di legarsi sia con l’acqua sia con la parete cellulare dell'epitelio, i colliri a
base di acido ialuronico hanno dimostrato di poter aumentano notevolmente la stabilità del film lacrimale nei casi di scarso film muciparo. Sono state testate concentrazioni dallo 0,1% all’1%, ma
sembra che i risultati terapeutici migliori si ottengano a basse percentuali.
polietilenglicole: È un agente viscosante con una buona azione aderente sull’epitelio e sulla fase acquosa. Sostituisce molto bene lo strato mucoide.
condroitin solfato: È una sostanza viscoelastica. La sua viscosità è inferiore a quella della metilcellulosa. Ma è più affine alla struttura cellulare epiteliale. Nei trattamenti di pazienti con CCS infatti, si sono ottenute risposte soggettive e oggettive migliori rispetto a quelle risultate dall’uso di colliri a base di acido ialuronico, alcool polivinilico, metilcellulosa.
effetti collaterali dei conservanti
Sempre più spesso i sostituti lacrimali vengono distribuiti in confezioni monodose per evitare l’uso di conservanti. I conservanti, infatti, si sono dimostrati irrispettosi nei confronti dei sostituti lacrimali (mantenimento e/o ricostruzione qualitativa del film lacrimale).
- belzanconio cloruro (sostanza cationica): a concentrazioni superiori allo 0,01% a livello corneale, provoca rottura dei ponti intercellulari dell’epitelio, danno ai microvilli, riduzione delle
cellule mucipare. La sua capacità di lesione dello strato lipido ne fa' un nemico della stabilità del film lacrimale.
- clorexidina (sostanza cationica): molti studi hanno confermato l'esistenza di un processo di allergizzazione da parte dell’occhio posto lungamente a contatto con la clorexidina. La sostanza
infatti tende a legarsi facilmente ai depositi lipido-proteico presenti sulle lenti morbide.
- clorobutanolo (alcali): l’atomo di cloro presente in questo conservante aumenta la solubilità dei lipidi e di conseguenza mina la stabilità dello strato lipidico.
- timerosal (composto mercuriale): non sono state registrate reazioni tossiche locali. Tuttavia molti pazienti possono manifestare ipersensibilità per la facilità di contatto con questo conservante. Infatti è molto diffuso perché è utilizzato anche in campi diversi dall’oftalmologia e dalla medicina.
preparazione di soluzioni isotoniche
E' accertato che i fenomeni osmotici hanno un'importanza rilevante nel mantenimento dell'omeostasi. Così, uno dei maggiori problemi nella pratica clinica è il mantenimento di adeguati fluidi corporei unitamente all'appropriato bilancio tra volumi di fluidi extra e intracellulari nei pazienti gravemente malati. Dovrebbe essere tenuto a mente, comunque, che variazioni fuori norma di fluidi ed elettroliti non costituiscono un disturbo, bensì sono la manifestazione di un disturbo.
Di séguito sono proposti alcuni esercizi esemplificativi per la prepazione di soluzioni isotoniche.
Il procedimento più comune è il metodo equivalente del cloruro di sodio: l'equivalente, E, in cloruro di sodio di un farmaco è definito come il peso necessario (di quel
farmaco) per produrre la stessa pressione osmotica dell'NaCl.
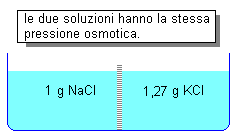 Per esempio, due soluzioni di cui una contenente 1 g di NaCl e l'altra 1,27 g di KCl, hanno circa la stessa pressione osmotica. |
Poiché le proprietà colligative sono realmente valide per soluzioni diluite di non elettroliti, i valori
degli equivalenti sono presenti in tabelle ottenute da dati sperimentali e non sono calcolati teoricamente basandosi sull'eguaglianza del numero di moli. Così, per esempio, 0,76 è l'equivalente
di KCl in cloruro di sodio.
|
esempio 1: rendere isotonica, per aggiunta di NaCl, una soluzione acquosa costituita da 60 ml di omatropina bromidrato 1% (E = 0,17); Per prima cosa, si calcola a quanto corrisponde in NaCl la soluzione di omatropina in questione:
quantità di omatropina bromidrato = 0,01 · 60 = 0,6 g Per avere una soluzione isotonica, il clururo di sodio deve essere presente in concentrazione di 0,9% Il NaCl totale richiesto per 60 ml è: 60 · 0,9/100 = 0,54 g Poiché i 60 ml di omatropina bromidrato all'1% è come se contenessero 0,102 g di NaCl, la quantità di NaCl ancóra da aggiungere = 0,54 - 0,102 = 0,438 g Se invece del NaCl si vuole aggiustare la tonicità con acido borico (E = 0,5), si ha: 1 g di acido borico = 0,5 NaCl di g, dunque i grammi di NaCl necessari (0,438) possono essere sostituiti da 0,876 g di acido borico |
|
esempio 2: rendere isotonica, per aggiunta di NaCl, una soluzione acquosa costituita da Efinefrina cloridrato (E = 0,3) in 30 ml di soluzione acquosa 0,5% e
Solfato di Zn (E = 0,15) in soluzione 0.3%
quantità di Efinefrina = 0,005 · 30 = 0,15 g
quantità di solfato dello zinco = 0,003 · 30 = 0,09 g Equivalente totale in NaCl = 0,045 + 0,0135 = 0,0585 g La quantità richiesta dal solo NaCl per rendere la soluzione isotonica = 0,009 x 30 = 0,27g Quindi, la quantità di NaCl ancóra da aggiungere per rendere la soluzione isotonica = (0,27 - 0,058)g = 0,212 g In alternativa, si può rendere la soluzione isotonica con acido borico (E = 0,5), aggiungendone 0,42 g |
|
esempio 3: rendere isotonica, per aggiunta di NaCl, una soluzione acquosa costituita da glucosio (E = 0,16) in 500 ml di soluzione acquosa 5% e cefaloridina (E =
0,07) in soluzione al 1%
quantità di glucosio = 0,05 · 500 = 25 g
quantità di cefaloridina = 0,01 · 500 = 5 g Equivalente totale in NaCl = 4 + 0,35 = 4,35 g La quantità richiesta dal solo NaCl per rendere la soluzione isotonica = 0,009 x 500 = 4,5 g Quindi, la quantità di NaCl ancóra da aggiungere per rendere la soluzione isotonica = (4,5 - 4,35)g = 0,15 g
|
|
esempio 4: verificare se è isotonica una soluzione costituita da Neomicina solfato (E = 0,11) in 100 ml di soluzione acquosa al 2%
Quantità equivalente di NaCl = 0,02 · 100 · 0,11 = 0,22g Quantità di NaCl richiesta per rendere la soluzione isotonica = 0,9/100 · 100 = 0,9 g In questo caso, mancando la compensazione del NaCl, la preparazione non è isotonica ma ipotonica. |
|
esempio 5: verificare se è isotonica una soluzione costituita bicarbonato di sodio (E = 0.65) in 60 ml di soluzione acquosa al 10%
Quantità di bicarbonato di sodio = 0,1 · 60 = 6g Quantità di quantità equivalente di NaCl = 6 · 0,65 = 3,9 g Quantità di NaCl richiesta per rendere la soluzione isotonica = 0,009 · 60 = 0,54 g Quindi, la soluzione non è isotonica ma è ipertonica |
metodo dell'abbassamento crioscopico
A volte, per un dato farmaco non sono disponibili i dati per l'equivalente in cloruro di sodio; in questo caso, si può utilizzare il metodo dell'abbassamento crioscopico.
|
esempio 1: rendere isotonica, per aggiunta di NaCl, una soluzione di desametazone sodio solfato 0,1% (0,5% Dt =
0,050 ºC ) in 30 ml di soluzione acquosa.
conoscendo l'abbassamento crioscopico per la soluzione di desametazone allo 0,5%, ammettendo una relazione lineare con la concentrazione, si può ricavare l'abbassamento crioscopico per una soluzione allo 0,1% 0,050 : 0,5% = x : 0,1% da cui si ricava x = Dt = 0,01 ºC si trova sperimentalmente che l'abbassamento crioscopico dell'acqua dovuto alla presenza di NaCl allo 0,9% è Dt = 0,52 ºC poiché una parte di NaCl sarà sostituita dal desametazone, per l'isotonicità dovrà aversi: Dt (per NaCl da solo) = 0,52 ºC = Dt (per NaCl) + 0,01 ºC (per desametazone) quindi, si dovrà addizionare alla soluzione di desametazone NaCl in quantità tale da ottenere un abbassamento crioscopico pari a 0,51 ºC conoscendo l'abbassamento crioscopico per la soluzione di NaCl allo 0,9%, ammettendo una relazione lineare con la concentrazione, si può ricavare la concentrazione necessaria per ottenere un abbassamento crioscopico pari a 0,51 ºC 0,9% : 0,52 = x : 0,51 da cui si ricava x = NaCl% = 0,88% i grammi di NaCl necessari per 30 ml di soluzione, sono dati dalla proporzione: 0,88 : 100 = x : 30 da cui si ricava x = 0,27 g NaCl Pertanto, la nostra soluzione isotonica conterrà 0,27 g NaCl e 0,03 g di desametazone. |
preparati per via inalatoria
L'uso di sostanze inalate nel trattamento delle malattie respiratorie è molto antico ed i suffumigi medicamentosi, ancóra praticati, con vapori di eucaliptolo o mentolo, sono la testimonianza di una pratica terapeutica che dura da secoli.
La terapia inalatoria è considerata la metodica di trattamento d'elezione nelle patologie respiratorie bronchiali, con particolare riferimento alla patologia asmatica acuta e cronica, in quanto permette di ottenere delle maggiori concentrazioni di farmaco in loco e minimizzare gli effetti collaterali rispetto ai farmaci assunti per via sistemica.
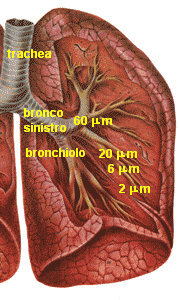
| fosse nasali, laringe e faringe | oltre 30 mm |
| trachea | 20-30 mm |
| bronchi e polmoni | 10-20 mm |
| bronchioli terminali | 3-10 mm |
| canali alveolari e alveoli polmonari | meno di 3 mm |
La via inalatoria è particolarmente utile per la somministrazione di farmaci gassosi o volatili (anestetici generali); ma anche i farmaci non volatili possono essere somministrati in forma di particelle altamente disperse, solide (polverizzate, aerosol solido) o liquide (aerosol liquido). La tabella in alto riporta il sito respiratorio raggiungibile a seconda della dimensione delle particelle disperse. La figura a sinistra, invece, riporta - con maggior immediatezza visiva - la dimensione massima approssimativa delle particelle che possono raggiungere le varie regioni del tratto respiratorio.
aerosol dosati (AD)
Questi piccoli dispositivi sono in commercio da molti anni sotto forma di bombolette pressurizzate di piccole dimensioni e di uso facile e veloce. Il farmaco è solubilizzato o sospeso nel propellente gassoso addizionato a piccole quantità di tensioattivo per impedire l'aggregazione delle particelle. L'aerosol vaporizzato dall'erogatore è una sospensione in aria di particelle solide e/o gocce di liquido. Tanto minore è la grandezza delle particelle tanto maggiore è la loro capacità di penetrazione nell'albero bronchiale. Il principio attivo è veicolato da un propellente gassoso da solo o in miscela con un altro.
|
1- agitare autoahaler, e poi sollevate la levetta grigia; 2- espirate lentamente, mettete il boccaglio dritto tra le labbra serrandole (facendo attenzione a non ostruire con le dita i fori per l'aria); 3- inspirate quanto più possibile attraverso la bocca. Non fermatevi quando udite click: continuate con un'espirazione veramente profonda; 3- trattenete il fiato per circa 10 sec., quindi espirate docemente; 4- abbassate la levetta grigia. |
|
L'efficienza degli AD è legata ad una sufficiente sincronizzazione tra erogazione del prodotto e inalazione, la cui velocità di flusso non deve essere superiore a 20-30L/min. In questo modo il
10-15% della dose erogata raggiunge le basse vie aeree con una dispersione dell'80% nel canale alimentare. Per ovviare all'inconveniente della sincronizzazione erogazione-inalazione, sono stati
prodotti particolari aerosol pressurizzati (Autohaler), dotati di un meccanismo valvolare che si attiva automaticamente con flussi inspiratori di 30 L/min. L'uso degli AD può determinare
l'insorgenza di effetti collaterali conseguenti all'eccessiva deposizione del farmaco nell'orofaringe (candidosi orale, disfonia da corticosteroidi, tosse).
Particolarmente per i bambini, l'utilizzo degli spray dosati comporta una serie di errori tecnici1
 distanziatore Babyhaler |
- dimenticare di scuotere lo spray prima dell'uso (54%);
- dimenticare di espirare prima di erogare lo spruzzo (66%);
- flettere il collo durante l'inalazione (17%);
- problemi di coordinamento tra erogazione e inalazione (58%);
- inalare troppo rapidamente (68%);
- trattenere il respiro meno di 7 secondi (52%);
- bloccare l'inalazione subito dopo l'erogazione (46%)
Per superare questi problemi, e contemporaneamente aumentare la quota polmonare del farmaco somministrato, sono stati introdotti i distanziatori, o camere distanziatrici (v. foto a dx), che presentano alcuni vantaggi:
- non è necessaria una fine coordinazione;
- rallentando la velocità dell'aerosol, si consente al propellente di evaporare: viene ridotta la velocità delle particelle, specie di quelle più voluminose, che non hanno effetti terapeutici e che si depositano sulle pareti del distanziatore, anziché in bocca e nelle alte vie respiratorie, portando ad un minor assorbimento sistemico del farmaco ed a minori effetti collaterali;
Il tipo di distanziatore "ideale" non è stato ancora individuato, ma è importante sottolineare che nei bambini di età inferiore a 3 anni è utile inserire una maschera tra il distanziatore e la
bocca, per evitare la dispersione del farmaco.
In commercio sono disponibili vari tipi di camere distanziatrici (Aerochamber-Markos, Volumatic, -Babyhaler-Glaxo, Ottoventi-Menarini, Jet-Chiesi). Con l’impiego di camere distanziatrici di
grosse dimensioni (750ml) è possibile quasi raddoppiare la quantità di principio attivo che raggiunge le vie aeree distali. Con il Babyhaler (figura sopra, a destra) è possibile somministrare i
farmaci anche a bambini molto piccoli (>6 mesi) nel corso di 5-10 atti respiratori.
Alcuni produttori di farmaci per AD hanno anche disponibili degli spaziatori specifici per i farmaci da loro prodotti. Un particolare distanziatore è stato proposto come compatibile con tutti i
AD. In realtà i dati clinici e sperimentali a sostegno di questa affermazione sono scarsi e in teoria ogni confezione AD + spaziatore richiederebbe una sperimentazione specifica. La variabilità
della quantità di aerosol erogato (conseguente all'uso di vari AD più spaziatore di grosso volume) è stata recentemente sottolineata in un recente studio che ha dimostrato un 36% di differenza
tra i vari AD.
Per il momento, se si vuole utilizzare un distanziatore, è preferibile scegliere, se disponibile, quello raccomandato dalla ditta produttrice del farmaco adoperato.
propellenti
Esistono sostanzialmente due tipi di formulazioni disponibili per i farmaci somministrabili con i nebulizzatori: le sospensioni e le soluzioni. Queste ultime sono da preferire in quanto il principio attivo è completamente disciolto nel solvente e costituisce un unico liquido omogeneo. Per alcuni farmaci non solubili o instabili in soluzione si ricorre alle formulazioni in sospensione, sistema eterogeneo in cui si può distinguere una fase dispersa di particelle solide insolubili ed una fase continua di solvente.
Quando un gas liquefatto viene introdotto e sigillato in un recipiente, si crea un equilibrio fra una sua frazione che evapora e l'altra che rimane in fase liquida dove si scioglie o disperde il prodotto da aerosolizzare. La fase gassosa, infatti, esercita la propria pressione contro le pareti del contenitore e sul liquido, forzandolo ad attraversare il tubicino di pescaggio connesso con la valvola d'uscita. Quando la valvola viene aperta, premendo sull'apposito tasto, la fase liquida fuoriesce finché si interrompe il flusso o si esaurisce la quantità di gas propellente.
Il propellene che ha sempre un punto di ebollizione molto inferiore alla temperatura ambiente, evapora rapidamente, rilasciando nell'aria un getto di particelle liquide, l'aerosol. In particolare, l'evaporazione del propellente deve avvenire nell'istante in cui l'aerosol raggiunge la superficie corporea interessata - non prima - depositandovi sopra il prodotto attivo.
I propellenti negli aerosol forniscono l'energia necessaria per espellere il contenuto dell'erogatore e - se non hanno anche la funzione di solvente e/o diluente - possono essere solubili o insolubili nella fase liquida.
-
propellenti gassosi insolubili nella fase liquida: la fase liquida deve occupare un volume non superiore al 50% dell'erogatore, in modo da lasciare spazio al gas insolubile.
La pressione del gas, all'interno del recipiente, segue con buona approssimazione la legge di Boyle per i gas ideali:
PV = costante a temperatura costante
questo significa che quando viene spruzzato il 50% del contenuto, raddoppia il volume a disposizione del gas e quindi la pressione dimezza (considerando il sistema a temperatura costante).Questo fatto comporta la necessità di riempire il recipiente con propellenti a pressioni di circa 600 kPa (1 kiloPascal = 9.869 10-3 atm).
-
propellenti gassosi solubili nella fase liquida: la solubilità del gas nella fase liquida permette riempimenti più elevati, che possono raggiungere circa l'80% della capacità
del contenitore. In questo caso, a differenza dei gas insolubili nel solvente, la caduta di pressione conseguente ad ogni erogazione, viene compensata dal progressivo passaggio in fase vapore
del gas disciolto. In queste condizioni, la pressione interna al recipiente rimane costante finché è presente il propellente.
-
propellenti con funzione veicolante: sono i propellenti costituiti da gas liquefatto, dove l'effetto aerosol si ottiene sfruttando il principio dell'evaporazione.
Un gas liquefatto contenuto in un contenitore chiuso, è in equilibrio con la sua fase gassosa e questo comporta che non è necessario avere pressioni elevate per compensare la riduzione di pressione conseguente alle erogazioni.
Prima dell'accordo di Montreal del 1987, che ha messo al bando i clorofluorocarburi, erano comunemente impiegati il difluorodiclorometano ed il triclorofluorometano in associazione [sono ancóra ammessi negli aerosol dosati destinati alla terapia dell'asma e della pneumopatia cronica ostruttiva, grazie alla deroga per usi essenziali, prevista nel regolamento della Comunità Europea n. 3093/94, sostituito da regolamento CE n. 2037/2000 del 29 giugno 2000]; attualmente sono sostituiti da idrofluoroalcani (HFA).
L'equivalenza tra erogatori CFC e HFA non è sempre definibile; comunque, l'evaporazione del propellente lascia delle particelle di diametro più piccolo rispetto ai CFC, aumentando potenzialmente la deposizione polmonare del farmaco.
erogatori di polveri
Rispetto agli AD, hanno il vantaggio di non necessitare di coordinazione tra erogazione e inalazione, di non
contenere propellenti e di avere un costo più contenuto. Gli svantaggi sono dovuti al fatto che necessitano di flussi superiori ai 60L/min (ad eccezione del turbohaler, che necessita solo di
20-30 L/min), che contengono lattosio (può provocare carie e irritazione delle vie aeree), e che la quantità erogata può variare in relazione alla igroscopicità o alla
umidità. Non possono essere usati in caso di attacco asmatico.
Farmaci erogati con questi dispositivi (v. illustrazioni d'uso) sono il DSCG
(Spinhaler), il Salbutamolo (Rothaler-Diskhaler), la Terbutalina e la Budesonide (Turbohaler), il Beclometasone, il Salmeterolo e il Fluticasone (Diskhaler).
nebulizzatori
Il principio attivo è veicolato da un liquido erogato per azione meccanica da un recipiente a pistone o munito di pompetta di gomma. L'erogazione con questi dispositivi realmente ecologici, ha lo svantaggio di non garantire erogazioni uniformi per quantità; così, l'impiego dei nebulizzatori è principalmente indicato per veicolare sostanze che non necessitano di erogazioni uniformi (per es., eucaliptolo).
Il fatto che gli aerosol siano veicolati da un propellente gassoso, presenta lo svantaggio di provocare un effetto irritante; d'altra parte, permettono di ottenere dosi uniformi rispetto a quelle ottenibili con i nebulizzatori. In realtà, questo vantaggio è discutibile in quanto si deve considerare che, sebbene l'uso degli inalatori sia apparentemente facile, molti pazienti li usano impropriamante (ad es. arrestando l'inspirazione appena il getto di farmaco raggiunge l'orofaringe). In pratica, solo il 10-15% del farmaco erogato con un singolo spruzzo raggiunge i polmoni; il resto si deposita sulle pareti della cavità orale e viene deglutito.
bioequivalenza dei farmaci inalati per aerosol
Negli ultimi anni, a causa del rapido aumento dei sistemi di inalazione proposti, si è resa necessaria la valutazione della bioequivalenza dei farmaci inalati, e questa è stata possibile grazie allo sviluppo di vari metodi (in vitro ed in vivo).
Con i metodi in vitro è stato possibile definire la potenza delle erogazioni e la equivalenza delle caratteristiche degli aerosol. Con i metodi in vivo (Scintigrafia polmonare con Tc marcato) è stato invece definita la deposizione del farmaco non solo nei polmoni, ma anche in altre
sedi (orofaringe, tratto gastroenterico) che vengono direttamente o indirettamente interessate durante la somministrazione. Con studi di questo tipo è stato possibile dimostrare, ad esempio, che
la deposizione polmonare di Budesonide e Terbutalina è doppia se si usa un sistema di inalazione turbohaler rispetto agli usuali spray pre-dosati.
Un altro tipo di valutazione in vivo è quello che si può attuare con studi clinici di comparazione dell'efficacia dei farmaci somministrati per aerosol. Questi studi sono però difficili per la
presenza di numerosi fattori confondenti e perché richiedono casistiche molto numerose. Alcuni studi effettuati finora - in parte criticabili per i motivi suddetti - che hanno valutato la
capacità broncodilatatrice del salbutamolo, hanno dimostrato significative variazioni fra i vari inalatori predosati nel produrre broncodilatazione. Anche per quello che riguarda l’uso degli
spaziatori la compatibilità con i vari AD non è stata ancóra definitivamente accertata ed anzi, l'uso di questi strumenti, pur essendo di utilità in molti casi, aggiunge un elemento di
complessità nella definizione della bioequivalenza.
ecologia e CFC
L'anno è il 2065. Quasi due terzi di ozono della Terra è distrutto, non solo sopra i poli, ma in tutto il mondo. Il drammatico buco dell'ozono scoperto nel 1980, è praticamente presente tutto l'anno, con un gemello sopra il Polo Nord. La radiazione ultravioletta, UV, che a medie latitudini cade su città come Washington, DC, è abbastanza forte da causare scottature in soli cinque minuti. Mutazioni del DNA da radiazioni UV superano il 500 per cento, con probabili effetti dannosi sulle piante, animali e esseri umani.
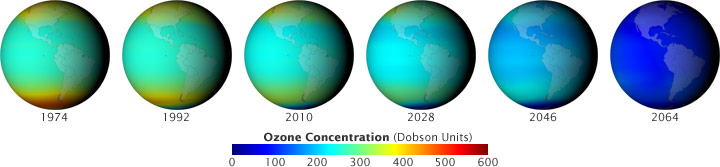
Questo sarebbe stato il futuro del mondo, se 193 nazioni non avessero accettato di vietare, attraverso il protocollo di Montreal 1989,l e sostanze chimiche dannose per l'ozono, secondo i chimici atmosferici della NASA Goddard Space Flight Center, dellaJohns Hopkins University, e la valutazione ambientale dei Paesi Bassi.
I problemi derivanti dai CFC, responsabili dell'assottigliamento dello strato atmosferico di ozono stanno portando ad una loro progressiva sostituzione con propellenti inerti e meno
dannosi.
L'evento negativo degli aerosol dosati, è rappresentato dall'evaporazione del propellente, attualmente della famiglia dei clorofluorocarburi, che contribuisce a modificare lo strato di ozono.
L'ozono (O3) è presente in tutta l'atmosfera.
Fino ad un'altitudine di 10 km (troposfera) la concentrazione di ozono è bassissima; si forma sia per eventi naturali (p.e. fulmini) sia per inquinamento atmosferico (autoveicoli, industrie): un
aumento della concentrazione di ozono può risultare dannoso per gli asmatici.
Ad altitudini maggiori 10-500 km vi è uno strato ad elevata concentrazione di ozono che rappresenta un filtro protettivo nei confronti delle radiazioni ultraviolette di tipo B i cui effetti
dannosi sono elevati (carcinomi della pelle, ustioni solari, fotosensibilizzazione, invecchiamento della pelle). La riduzione di questo strato di ozono è principalmente dovuta a composti volatili
a base di cloro e bromo.
Gli sforzi delle industrie volte a sostituire questi composti sono quindi giustificati e, per sollecitarne la sostituzione con prodotti meno nocivi, nel 1987 rappresentanti di varie nazioni hanno
sottoscritto un protocollo, detto di Montreal, che prevede una graduale messa al bando di questi composti. Poiché i nostri asmatici sicuramente subiscono un danno dall'inquinamento atmosferico
non c'è che da applaudire ed incoraggiare questi sforzi che proteggono il nostro ecosistema.
Ma a questo punto sorge spontanea la domanda: può il paziente asmatico essere considerato un "inquinatore"?.
Nel mondo si calcola che esistono circa 100 milioni di
asmatici; circa la metà di questi pazienti utilizzano le bombolette pressurizzate conteneti clorofluorocarburi ed ogni anno vengono fabbricati circa 440 milioni di bombolette.
Il mercato è grande, ma in termini generali si calcola che la quota di "inquinanti" utilizzata dai nostri pazienti corrisponde a circa 0,1 % della produzione totale di CFC: come dire che se si
anticipasse di un anno la sospensione di CFC per uso non terapeutico, i nostri pazienti potrebbero produrre un equivalente effetto inquinante in 1000 anni.
Questo significa che se anche improvvisamente interrompessimo l'uso delle bombolette il contributo ecologico sarebbe pressocché trascurabile.
Comunque, altre possibilità per evitare i CFC, possono essere rappresente da somministrazioni alternative (Inalatori di polveri, nebulizzatori meccanici e farmaci somministrabili per via orale.
apparecchi
In commercio sono attualmente disponibili diversi modelli di apparecchiature essenzialmente riconducibili a
due tipologie: nebulizzatori pneumatici ad aria compressa od ossigeno compresso, e nebulizzatori ad ultrasuoni. Questi ultimi hanno il vantaggio di una minor rumorosità ed una maggiore rapidità
di nebulizzazione, ma emettono particelle di dimensioni non omogenee e più grandi, che si depositano più centralmente nell'albero respiratorio; costano di più e si rompono con maggior facilità.
Pertanto dovrebbero essere consigliati solo quando è necessaria una somministrazione singola o ripetuta di quantità elevate di liquido in tempi brevi.
I nebulizzatori pneumatici producono particelle più omogenee, sono in genere meno costosi e sono più
resistenti all'uso.
I nebulizzatori sono molto usati nella terapia dell'asma e nei pazienti affetti da infezioni delle vie respiratorie. La somministrazione dei farmaci per via inalatoria con nebulizzatori è
particolarmente indicata nei bambini piccoli, che fisiologicamente hanno una respirazione superficiale e che quindi ad ogni atto respiratorio introducono solo un piccolo volume di aria. In questi
piccoli pazienti gli spray dosati o le bombolette pressurizzate o gli inalatori pre-dosati non possono perciò essere usati, anche perché richiedono una perfetta coordinazione tra l'erogazione del
farmaco e la sua inalazione.
L'aerosolterapia è inoltre una scelta obbligata quando è necessario somministrare per via aerosolica prodotti non disponibili in spray dosati come ad esempio antibiotici, mucolitici ecc.
Per penetrare nei bronchi i farmaci devono essere disintegrati in minuscole particelle: questo processo si verifica nella camera di nebulizzazione dell'apparecchio per aerosolterapia. Affinché un nebulizzatore possa considerarsi efficiente occorre che:
- produca particelle di diametro compreso tra 1 e 5 mm, cioè sufficientemente piccole da poter penetrare alla periferia dell'albero bronchiale (le particelle di dimensioni inferiori ad 1 mm penetrano molto in profondità nelle vie aeree, ma si depositano in bassa percentuale sulle pareti dei bronchi e vengono in gran parte espulse con l'espirazione mentre le particelle di diametro superiore ai 5 mm non penetrano in quantità significative al di sotto della laringe, depositandosi soprattutto a livello naso-faringeo);
- abbia una notevole gittata, sia in altre parole in grado di produrre grossi volumi di aerosol in tempi brevi ;
- produca particelle di dimensioni omogenee.
Affinché l'aerosol sia efficace la respirazione deve avvenire per via orale poiché il naso funge da filtro
trattenendo gran parte dell'aerosol che non può di conseguenza arrivare ai bronchi: attenzione quindi quando si usa il boccaglio che il bambino non respiri attraverso il
naso.
Riassumendo si può concludere che nell'acquisto di un apparecchio per aerosol dovrebbe essere controllate le
seguenti caratteristiche:
- ampolla di vetro o in materiale sintetico inalterabile;
- potenza prodotta superiore ad un'atmosfera;
- flusso d'aria non inferiore ai 6 L/min.;
- tempo di erogazione di una soluzione di 2-3 ml inferiore ai 10 minuti;
- produzione di particelle di diametro omogeneo compreso tra 1 e 5 micron.
L'esecuzione ottimale dell'aerosolterapia (nel senso di una migliore deposizione del farmaco nelle vie aeree più periferiche) deve rispettare le seguenti condizioni:
- deve essere usato preferibilmente il boccaglio (la mascherina riduce notevolmente la quantità di farmaco che raggiunge il polmone);
- deve essere esclusa la respirazione nasale;
- l'erogazione deve essere eseguita a volume corrente;
- il volume totale della soluzione da inalare deve essere di circa 3 ml se si usano nebulizzatori pneumatici e di 5 ml se si usano nebulizzatori ad ultrasuoni (tenere presente che la dose di farmaco che raggiunge i polmoni è il 10% circa di quella erogata dallo strumento alla bocca); la pressione di alimentazione ottimale è di 6L/min;
- l'erogazione della soluzione deve avvenire un periodo ininterrotto di tempo compreso tra i 5 e i 10 minuti. Un aumento del tempo di erogazione è indice di malfunzionamento, così come un aumento della rumorosità della pompa di compressione.
1(tratto da: Pedersen S, Frost L, Arnfred T. Errors in inhalation technique and efficiency in inhaler use in asthmatic children. Allergy 1986;41:118 – 124)
somministrazione topica nasale
Per la somministrazione topica di farmaci a livello nasale possono essere anche utilizzati i nebulizzatori pneumatici ad ultrasuoni muniti di appositi beccucci nasali. Da tenere presente che questo tipo di aerosolizzazione prevede particelle di piccolo diametro (1-8 mm) e che quindi può essere vantaggiosamente utilizzata quando si vuole cercare di raggiungere i seni paranasali (la deposizione di piccole particelle nei seni paranasali è possibile solo se il diametro degli osti è superiore ad un millimetro).
In generale però, per la terapia nasale si preferisce la somministrazione in gocce tramite appositi contagocce, la somministrazione tramite micropompe nebulizzatrici, e la somministrazione
tramite bombolette pressurizzate pre-dosate. La somministrazione in gocce permette (dato che di solito il farmaco è a bassa concentrazione) di applicare grandi volumi di liquido con una discreta
deposizione sulla mucosa nasale. D'altra parte, il dosaggio somministrato non è preciso. I nebulizzatori a micropompe sono ben tollerati e assicurano una buona deposizione sulla mucosa nasale, ma
anche per essi non è possibile controllare precisamente il dosaggio.
Gli spray pre-dosati assicurano un dosaggio preciso, ma presentano l'inconveniente di non distribuirsi uniformemente sulla mucosa nasale, e a causa del getto piuttosto violento, di poter causare
fastidio e quindi rifiuto della terapia. Recentemente sono stati anche proposti apparecchi a flusso forzato (Rinoflow) che assicurano un buon lavaggio nasale ma non migliorano la
deposizione nei seni paranasali.
preparazioni suppositorie
Con il termine suppositori, si intendono le forme farmaceutiche di consistenza solida o molle, destinate
all'introduzione rettale, vaginale o uretrale.
la F.U. prevede per le supposte e gli ovuli la determinazione dell'uniformità di massa, dell'uniformità di
contenuto (se il contenuto in p.a. è inferiore a 2 mg o inferiore al 2% della massa totale) della massa o volume rilasciabile, del tempo di dissoluzione.
supposte
L'ampolla rettale contiene un liquido, il cui pH (pH = 7.2), non essendo tamponato, varia in funzione della natura dell'eventuale medicamento introdotto. Le mucose rettali sono riccamente vascolarizzate dalle vene emorroidarie inferiori, medie e superiori; le prime due conducono direttamente nel circolo generale, mentre le vene superiori attraversano il fegato. In realtà, esperienze cliniche hanno dimostrato che buona parte del farmaco introdotto nell'ampolla rettale raggiunge comunque il fegato. I vantaggi di questa formulazione sono dunque riassumibili nella facilità di somministrazione ai bambini, nell'evitare l'inattivazione dei succhi gastrici ed in un buon assorbimento.
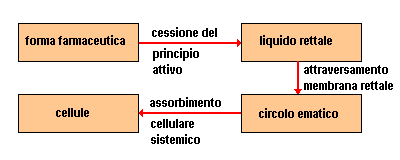
preparazione delle supposte
Il primo punto da considerare, è la scelta dell'eccipiente, necessario per raggiungere il volume necessario
per produrre le singole supposte; in questo esempio, ci riferiremo al burro di cacao.
Il secondo punto, è il cosiddetto "fattore di spostamento" o, più propriamente, "fattore di sostituzione", la
cui conoscenza è essenziale per calcolare la quantità di eccipiente da impiegare per preparare una supposta di dato peso.
 1 g di farmaco occupa un volume differente da quello occupato da 1 g di eccipiente: è quindi necessario tener conto del fattore di sostituzione. |
Posto che si vogliano preparare supposte contenenti ciascuna 0.5 g di cloralio idrato (sedativo ipnotico), utilizzando uno stampo per 6 supposte, con portata di 2 g per supposta (per uno specifico eccipiente), la quantità di eccipiente da utilizzare non sarà Qe = 12 - 6 · 0.5 = 9 , in quanto il principio attivo pur mescolandosi intimamente con l'eccipiente, occuperà un volume diverso (v. fig. a destra) Così, il calcolo corretto è dato dalla formula:
| (1) Qe = Qs - (Qp · f) |
dove:
Qs = portata dello stampo;
Qp = quantità di principio attivo;
f = fattore di sostituzione corrispondente alla quantità di eccipiente, espressa in grammi, sostituita da 1 g di principio attivo.
Nel caso proposto, f = 0,15 , pertanto applicando la eq. 1, si ha: Qe = 12 - 6 · 0,5 · 0,15 = 11,55
I fattori di sostituzione sono riportati su apposite tabelle, però possono essere determinati sperimentalmente.
Posto, ad es., che si voglia calcolare il fattore di sostituzione di un farmaco A:
- si prepara una miscela di 20 g di cui il 20% è A (16 g eccipiente, 4 g farmaco);
- si cola la miscela in uno stampo la cui portata, per un dato eccipiente, sia nota (ad es. 12 g);
- si pesano le supposte ottenute: ad esempio, 14 g;
- si calcola il quantitativo di A presente nelle supposte ottenute (14 · 0,20 = 2,8 g)
- si calcola per differenza l'eccipiente effettivamente utilizzato (14 - 2,8 = 11,2)
i dati disponibili sono: Qp = 2,8 g ; Qe = 11,2 g ; Qs = 12 g
ricavando f dalla eq. 1, si ottiene: f = (12 - 11,2)/2,8 = 0,29
Si ponga attenzione all'importanza del fattore di
sostituzione. Il riempimento delle valve avviene in modo volumetrico; quindi, ognuna di esse deve contenere la quantità di farmaco richiesta. Se si ragiona ponderalmente e non si
considera f, la quantità versata nelle valve dovrà essere poi portata a volume con eccipiente. Infatti, abbiamo visto che con f = 0.15 la quantità di eccipiente
da utilizzare è 11.5 g e non i 9 g calcolati ponderalmente.
Esempio: calcolare la quantità di eccipiente necessaria per allestire 10.000 supposte da 2 g contenenti ciscuna 0,5 g di cloralio idrato.
Il calcolo è immediato: 2 × 10.000 - 0,5 × 10.000 = 15 Kg di eccipiente;
considerando il fattore di sostituzione: 2 × 10.000 - 0,5 × 10.000 × 0,15 = 19,25 Kg di eccipiente. Come si vede, con un riempimento volumetrico, se non si considera il fattori di sostituzione,
la quantità di eccipiente calcolata non permette di preparare il numero di supposte richiesto.
 stampo metallico per supposte. Prima della colatura, lo stampo viene chiuso con due viti a testa zigrinata. |
Il terzo punto da tenere in considerazione è l'abbassamento del punto di fusione conseguente all'aggiunta di un farmaco solubile nell'eccipiente. Posto che si vogliano preparare supposte con 1,46 g di burro di cacao e 0,5 g di cloralio idrato, possiamo facilmente calcolare l'abbassamento crioscopico:
| Dt = K c |
dove:
K = coefficiente crioscopico (K burro di cacao = 10);
c = concentrazione molale (moli di soluto/kg di solvente)
Nell'esempio proposto, il P.M. del cloralio idrato è 165 e quindi, c = 0,5 · 1000/(165 · 1.46)= 2,07; inserendo questo valore nella eq. 2, si trova t = 20,7 ºC. Per compensare l'abbassamento crioscopico (che può produrre un rammollimento della supposta), si può aggiungere cera, in quantità non inferiore al 6%
La scelta dell'eccipiente è quindi molto importante. Come principio generale il p.a. deve essere insolubile o poco solubile nell'eccipiente. Ciò per evitare l'abbassamento del punto di fusione della supposta e per evitare che il p.a., in virtù di un elevato coefficiente di ripartizione, non venga rilasciato facilmente dalla base.
Per la produzione industriale delle supposte, la massa grassa viene fusa ed addizionata ad altri eccipienti ed al principio attivo; la miscela ottenuta viene omogeneizzata e mantenuta in sospensione tramite un agitatore e quindi trasferita in modo automatico alle linee di confezionamento. I contenitori di alluminio (valve) che racchiudono le supposte, vengono appositamente prodotti tramite la stampa e la saldatura di due fogli di alluminio. Nelle valve appena forate viene iniettata a pressione la sospensione proveniente direttamente dall'impianto di preparazione. Il raffreddamento avviene in modo graduale; le valve vengono sigillate, tagliate e inviate all'astucciatrice.
 Saas 15 - macchina automatica per la produzione di supposte in alveoli di alluminio e di materiale plastico termoformabile (brevetto Sarong - www.sarong.it). |
ovuli vaginali
Contrariamente alle supposte, questa forma farmaceutica è destinata a svolgere esclusivamente un'azione locale a livello della mucosa vaginale e dell'utero. La ricca vascolarizzazione della mucosa vaginale rende comunque possibile l'assorbimento di farmaci e ciò può portare ad indesiderati quanto imprevedibili fenomeni di ordine generale. A questo proposito, ha fondamentale importanza la scelta dell'eccipiente: deve consentire una cessione del farmaco lenta e continua, in modo da mantenere, a livello della mucosa interessata, una concentrazione che garantisca per un tempo adeguato l'effetto terapeutico richiesto.
Un eccipiente che fonde rapidamente (eccipienti lipofili per supposte), e rilascia globalmente il farmaco, non è adatto in quanto la massa liquefatta verrebbe drenata dalla secrezione vaginale senza permettere al farmaco di rimanere a contatto con la mucosa per un tempo sufficiente; inoltre, potrebbe produrre effetti indesiderati o dannosi qualora il farmaco fosse rapidamente ed intensamente assorbito.
Gli eccipienti ottimali per gli ovuli sono pertanto quelli che non fondono alla temperatura del corpo, ma si dissolvono gradualmente nella secrezione vaginale, assicurando così un apporto di
farmaco continuo e controllato, via via che questo viene drenato o assorbito. Un classico eccipiente è la gelatina glicerinata: gelatina (10 parti), acqua depurata (25 parti), glicerina (65
parti); si possono variare le proprietà di questo eccipiente a seconda dei farmaci che vi vengono incorporati. Ciò allo scopo di adattare la velocità con la quale il farmaco deve essere liberato
e per tener conto della sua possibile influenza sulle modalità di gelificazione e quindi sulla consistenza della massa.
Come eccipienti per ovuli possono essere impiegate anche miscele di polietilenglicoli con
caratteristiche fisiche diverse ed in varie proporzioni, le cui diverse velocità di dissoluzione nella secrezione vaginale si possono adattare alla velocità con cui i farmaci devono essere
rilasciati per le diverse esigenze terapeutiche.
Poiché gli ovuli sono destinati ad un'azione locale, è accettabile una differenza nel contenuto unitario di farmaco in ciascun ovulo. Pertanto, a differenza delle supposte per le quali si considera il fattore di sostituzione, la massa, preparata alla necessaria concentrazione di sostanze attive, va direttamente colata nello stampo ripartendola in ovuli di dimensioni opportune.
preparazioni dermatologiche
Le preparazioni destinate ad uso locale sono: unguenti, pomate, creme, lozioni, gel, schiume.
- unguenti: costituiti da una base a basso contenuto di acqua, sono indicati nel trattamento delle dermatosi secche, squamose ad impronta lichenoide ed ipercheratosica. La base grassa, per le sue proprietà occlusive, esplica infatti un vantaggioso effetto emolliente che permette al farmaco di agire in profondità;
- pomate: costituite da una base grassa contenente una modica percentuale di acqua, sono indicate, in qualunque localizzazione, nel trattamento delle superfici cutanee delicate, sia secche, sia umide, purché non eccessivamente secernenti. In particolare, le pomate assicurano alla cute un appropriato apporto lipidico, senza bloccare la respirazione e gli scambi di calore.
-
creme: costituite usualmente da emulsioni O/A poco grasse, sono particolarmente indicate nel trattamento di lesioni secernenti e di zone cutanee umide, come la regione
inguinale ed il cavo ascellare, dove è opportuno impiegare una base con elevato contenuto acquoso che permetta il flusso del secreto ed induca una rapida essudazione ed essiccamento della
cute.
- lozioni: sono indicate quando si debbano applicare piccole quantità su vaste superfici cutanee, o per applicazione in zone di difficile accesso quali quelle pelose.
- gel: sono indicati nelle forme essudative, o quando sia desiderato per localizzazioni particolari un veicolo trasparente.
- schiume: sono impiegate per ottenere un'azione detergente medicata della cute glabra o pelosa, delle unghie infette da miceti, lieviti, ecc. Offrono il vantaggio di sostituire il sapone che potrebbe irritare la pelle già compromessa dall'infezione.
classificazione delle preparazioni dermatologiche secondo la F.U. XI
Secondo la F.U., le pomate non sono definite e vengono implicitamente suddivise in unguenti e creme, comprese tra le preparazioni semisolide per applicazione cutanea.
Le preparazioni semisolide per applicazione cutanea sono destinate al rilascio locale o transdermico di princìpi attivi, oppure hanno azione emolliente o protettiva. Hanno aspetto omogeneo.
Le preparazioni semisolide per applicazione cutanea sono costituite da una base semplice o composta in cui, usualmente, sono disciolti o dispersi uno o più princìpi attivi. Secondo la sua
composizione, la base può influenzare l'azione della preparazione.
Le basi possono essere costituite di sostanze naturali o sintetiche e possono essere sistemi a una fase o multifase. Secondo la natura della base la preparazione può avere carattere idrofilo o
idrofobo (lipofilo), può contenere additivi adatti come antimicrobici, antiossidanti, stabilizzanti, emulsionanti, addensanti e sostanze che aumentano l'assorbimento.
Le preparazioni destinate all'uso su larghe ferite aperte o su pelle gravemente danneggiata sono sterili.
Si possono distinguere varie categorie di preparazioni semisolide per applicazione cutanea:
- unguenti;
- creme;
- geli;
- paste;
- cataplasmi;
- impiastri medicati
Secondo la loro struttura, unguenti, creme e geli generalmente hanno un comportamento viscoelastico ed un carattere non newtoniano, presentando un flusso plastico, pseudoplastico, dilatante (le paste); tutte queste preparazioni possono presentare caratteristiche tixotropiche.
unguenti: un unguento è costituito da una base monofasica in cui possono essere disperse sostanze solide o liquide.
- unguenti idrofobi (lipofili): possono assorbire solo piccole quantità di acqua. Tipiche sostanze usate per la loro preparazione sono paraffine solide, semisolide e liquide, oli vegetali, grassi animali, gliceridi sintetici, cere e polialchilossilani liquidi.
- unguenti che emulsionano acqua: possono assorbire maggiori quantità di acqua e formare perciò emulsioni acqua in olio (A/O) oppure emulsioni olio in acqua (O/A) secondo la natura dell'emulsione: a questo scopo si possono usare agenti tensioattivi A/O come alcoli della lana, esteri del sorbitano, monogliceridi e alcoli grassi oppure emulsionanti O/A come solfati di alcoli grassi, polisorbati, macrogol cetostaril esteri o esteri di acidi grassi con magrogol. Le loro basi sono quelle degli unguenti idrofobi.
- unguenti idrofili: sono preparazioni che hanno basi miscibili con l'acqua. Le basi sono miscele di macrogol (polietilenglicoli) liquidi e solidi. Possono contenere appropriate quantità di acqua.
creme: le creme sono preparazioni multifase costituite da una fase lipofila e da una fase acquosa.
- creme idrofobiche: hanno come fase continua la fase lipofila. Contengono emulsionanti acqua in olio (A/O) come alcoli della lana, esteri del sorbitano e monogliceridi.
- creme idrofile: hanno come fase continua la fase idrofila. Contengono emulsionanti olio in acqua (O/A) come saponi di sodio e trietanolammmina solfati di alcoli grassi, polisorbati e acidi grassi poliossidrilati ed esteri di acidi grassi associati, se necessario, con emulsionanti acqua in olio (A/O)
gel: sono costituiti da liquidi gelificati con opportuni gelificanti.
- gel idrofobi (oleogel): sono preparazioni le cui basi usualmente sono costituite da paraffina liquida con polietilene od oli grassi gelificati con silice colloidale o saponi di alluminio o di zinco.
- gel idrofili (idrogel): sono preparazioni le cui basi solitamente contengono acqua, glicerolo o glicole propilenico, gelificati con adatte sostanze come amido, derivati dalla cellulosa, polimeri carbossivinilici e silicati di magnesio alluminio.
paste: sono preparazioni semisolide per applicazioni cutanee che contengono, finemente dispersi nella base, solidi in grandi proporzioni.
cataplasmi: costituito normalmente da impasti di amidi, mucillagini e oli, in cui sono dispersi princìpi attivi solidi o liquidi. Si applicano, possibilmente a caldo, direttamente sulla pelle.
impiastri medicati: sono preparazioni flessibili (cerotti medicati) che contengono uno o più princìpi attivi. Sono destinati ad uso topico e sono preparati per mantenere i
princìpi attivi in stretto contatto con la pelle così che possono essere assorbiti lentamente oppure agire come protettivi o cheratolitici.
Gli impiastri medicati sono costituiti da una base adesiva, che può essere colorata, contenente uno o più princìpi attivi, spalmata come strato uniforme su un appropriato supporto fatto di
prodotti naturali o sintetici. Non è irritante o sensibilizzante per la pelle. Lo strato adesivo è protetto con un adatto rivestimento che si toglie prima di applicare l'impiastro alla pelle.
Quando si toglie, lo strato protettivo non deve staccare la preparazione dello strato esterno di sostegno. Gli impiastri medicati sono presentati in una varietà di misure adatte al loro uso
previsto oppure in fogli più grandi da tagliare prima dell'uso. Aderiscono saldamente alla pelle applicando una lieve pressione e possono essere tolti senza causare danno apprezzabile.
![]()
schiume medicate: sono preparazioni costituite da grandi volumi di gas disperso in un liquido generalmente contenente uno o più princìpi attivi, un tensioattivo che assicuri
la loro formazione e vari altri eccipienti. Sono di solito destinate ad essere applicate sulla cute o sulle mucose.
Le schiume medicate si formano generalmente al momento della somministrazione da una preparazione liquida contenuta in un contenitore pressurizzato, dotato di un dipositivo costituito da una
valvola e da un tasto a pressione, adatto per l'erogazione della schiuma.
Le schiume medicate destinate ad essere impiegate su pelle gravemente lesa e su ferite aperte, sono sterili.
classificazione delle basi per uso dermatologico
la classificazione delle basi è correlata alle loro caratteristiche chimico fisiche: si possono avere basi idrofile o idrofobe, sia in forma monofasica che emulsionata.
Gli eccipienti lipofili monofasici sono sostanze non emulsionate che possono essere distinte in:
- basi che non hanno alcuna affinità per l'acqua (grassi, sugna benzoinata, vaselina, spermaceti, siliconi, ecc.). Sono utilizzate quando occorre preparare unguenti con sostanze che restano in sospensione, oppure di natura grassa.
- basi che, se anidre, hanno una certa affinità per l'acqua e le soluzioni acquose (lanolina, alcoli della lana, ecc.). Quando non sono più anidre, assumono la caratteristica di sostanze bifasiche. La quantità massima di acqua che può essere trattenuta da queste sostanze si definisce "numero d'acqua" e corrisponde ai grammi di acqua che possono essere trattenuti da 100 g della sostanza alla temperatura di 20 ºC per almeno 24 ore.
Gli eccipienti idrofili monofasici sono generalmente sostanze ad elevato P.M., tipo i PEG (polietilenglicoli), i gel, ecc.
Gli eccipienti idrofili bifasici sono costituiti da:
- emulsioni A/O in cui il principio attivo può essere solubile sia nella fase acquosa che in quella oleosa. Quando queste creme vengono applicate, si ha una certa evaporazione della fase acquosa con conseguente sensazione di fresco (da cui il nome:cold cream). Trattandosi di preparazioni emulsionate, occorrerà stabilizzarle con i metodi discussi. Inoltre, può essere necessario utilizzare i conservanti per prevenire i problemi che possono derivare da un eventuale irrancidimento della fase esterna.
- emulsioni O/A in cui il principio attivo può essere solubile sia nella fase oleosa che in quella acquosa; quest'ultima può far sorgere il problema di un inquinamento da microorganismi e può essere necessario utilizzare un conservante.
tecniche di preparazione
Industrialmente le preparazioni solide vengono lavorate utilizzando apposite impastatrici. In alcuni casi è
necessario operare termicamente per mantenere i componenti allo stato fluido. Tutte queste preparazioni richiedono una preventiva setacciatura delle polveri in modo che nel prodotto non siano
presenti granuli abrasivi. Si può comunque raffinare il preparato facendolo passare in una raffinatrice a cilindri (v. figura a sinistra): consiste di tre cilindri rotanti, parallelati in modo
che la distanza tra il primo e secondo rullo sia inferiore a quella tra il secondo ed il terzo; la velocità di rotazione dei rulli aumenta da sinistra verso destra. L'azione di riduzione delle
particelle è dovuta sia all'azione combinata di compressione, prodotta dai rulli, sia allo stiramento generato per effetto delle diverse velocità di rotazione.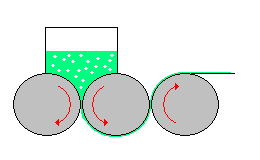
Nel caso delle emulsioni, occorrerà eomogeneizzare il tutto in modo da ridurre il più possibile le dimensioni delle particelle e sfavorire quindi il processo di separazione delle fasi. Per questo
scopo, si usano appositi omogeneizzatori.
cold cream
Si fa risalire a Galeno la preparazione di una crema composta dalla fusione di cera d'api, nella quale, dopo aver aggiunto 3 parti di olio d'oliva nel quale erano stati messi in infusione petali di rosa, veniva incorporata la maggior quantità d'acqua possibile. Questa prerparazione fu inclusa nella Farmacopea Londinese (1618), e nel 1914 venne modificata con l'aggiunta di una piccola quantità di borace. La particolarità di questa crema, pur essendo a base grassa, è la notevole azione rinfrescante dovuta all'evaporazione dell'acqua. Per questa ragione, le venne dato il nome di cold cream (in effetti, tutte le creme e pomate hanno una certa azione rinfrescante, ma il nome cold cream è riferito specificamente a questa preparazione).
La crema formulata secondo la ricetta in tabella a destra, è un'emulsione A/O bianca, lucida, priva di granulosità e facilmente spalmabile. La sua particolarità è costituita dalla presenza del borace che idrolizzandosi secondo lo schema di reazione in basso, libera degli ioni ossidrile che neutralizzaano gli acidi grassi liberi (presenti nella cera d'api) formando così un sapone alcalino che funziona da emulsionante: dunque il tensioattivo non viene aggiunto in quanto si produce durante la formazione dell'emulsione. La quantità di borace è all'incirca il 6% della cera d'api. La preparazione viene effettuata a caldo in modo da fondere la cera ed a questa si addiziona l'olio e l'acqua contenenete il borace.
| cera d'api | 20.0 g |
| olio minerale | 50.0 g |
| borace | 12.0 g |
| acqua, aromatizzante | q.b. a 100 g |
2[B(OH)4] - ![]() 2
H3BO3 + 2OH -
2
H3BO3 + 2OH -
due tipiche preparazioni dermatologiche a freddo
| potassio ioduro | g 2,50 |
| sodio tiosolfato | g 0,10 |
| vaselina q.b. a | g 30,0 |
| S. per applicazioni locali | |
Questa preparazione si esegue solubilizzando i due sali nella minima quantità d'acqua
necessaria. Poiché la paraffina (vaselina) è idrofoba, si fà assorbire la soluzione salina ad una analoga quantità di lanolina anidra. Successivamente si addiziona la vaselina mescolando fino a
completa omogeneizzazione.
| zinco ossido | |
| olio di vaselina | anag 3 |
| vaselina filante | g 24,0 |
| S. per applicazioni locali | |
Questa preparazione è simile al prodotto commerciale pasta di Fissan® : è indicata per proteggere con una pellicola protettiva la pelle del bambino dall'umidità presente nel pannolino bagnato. In questo caso, l'ossido di zinco (che fornisce il componente del film protettivo) non è solubile in acqua e quindi, a differenza della preparazione precedente, lo si deve incorporare direttamente nella vaselina. Prima, però, con una spatola si leviga su piastra di marmo l'ossido di zinco lubrificandolo con una pari quantità di olio di vaselina (paraffina liquida), poi si miscelano la vaselina liquida e la vaselina filante.
 Time in Rome
Time in RomeSOPRAVVIVERE FUORI CASA
Studenti ai fornelli

Un ebook per sopravvivere alla vita da universitario o lavoratore fuori casa
Decisamente carino.....
- cercare una stanza
- sopravvivere senza la cucina di mamma
- Vita da studente
i principali argomenti, che trovo utili anche per quanti di noi si spostano per i primi incarichi ad esempio




